INTRODUCTION
Cardiovascular disease (CVD) is the leading cause of death in the United States, Europe and Japan [1] and is poised to become the most significant health problem worldwide. According to the World Health Organisation (WHO), an estimated 17 million people die of CVDs, particularly heart attack, stroke and heart failure, every year. In Mauritius deaths due to cardiovascular dysfunctions have kept on increasing for the last 10 years attaining the 51% mark in 2004 [2]. Cardiovascular disease is of multifactorial etiology associated generally to a variety of risk factors for its development including hypercholesterolaemia, hypertension, smoking, diabetes, poor diet, stress and physical inactivity amongst others. During the last few decades, research data has prompted a passionate debate as to whether oxidation, or specifically, oxidative stress mediated by free radicals/reactive oxygen species (ROS)/reactive nitrogen species (RNS), is a primary or secondary cause of many chronic diseases. As a result, scientific resources have focused to a large extent on the role that antioxidants could play to delay or prevent oxidative stress and consequently the incidence of chronic disorders. This article will review the biology of ROS/RNS, their pathways through which they relate to the pathology of cardiovascular disease. We shall also discuss the roles that antioxidants may play in controlling oxidative stress and reduce the incidence of CVDs.
BASIC CONCEPTS OF FREE RADICALS, REACTIVE NITROGEN SPECIES, REACTIVE OXYGEN SPECIES AND OXIDATIVE STRESS
A free radical is any chemical species (capable of independent existence) possessing one or more unpaired electrons, an unpaired electron being one that is alone in an atomic or molecular orbital. Free radicals are formed from molecules via the breakage of a chemical bond such that each fragment keeps one electron (free radicals may also be formed by collision of the non radical species by a reaction between a radical and a molecule -which must then result in a radical since the total number of electrons is odd), by cleavage of a radical to give another radical and, finally via redox reactions [3-5]. Radicals are generally less stable than non-radical species, although their reactivity varies (Table 1).
Unimolecular Radical Reactions Reactions result from the instability of the first formed radical. The radicals may completely decompose or rearrange before reaction with other molecules or radicals present. Radical-Molecule Interactions Addition to unsaturated systems AR — X + Y. —> AR — Y + X. Abstraction or displacement: SH2 reactions‡ Ph. + CBrCl3 —> .CCl3 + PhBr - Homolytic substitution at multivalent atoms also occurs but both do not normally occur at saturated carbon centers. Reaction with oxidizing agents Ph.CHCHRCO2Et+ Cu2+ —> PhC+HCHRCO2Et + Cu+ Radical-Radical Interactions Dimerization or radical coupling R’. + R”. —> R’ -R” (When R’ = R”, the reaction is dimerization and when R’≠ R” the reaction is radical coupling or combination) Radical disproportionation |
| †The reader is referred to Moad and Solomon [5] for an extensive overview of the reaction sequences highlighted in this table and an examination of the complex chemistry of organic radicals. ‡SH2 stands for substitution homolytic biomolecular. ØThe disproportionation reaction derives its driving force from the formation of two newstrong bonds and from the fact that the b-CH bonds in radicals are usually weak. |
Free radicals and reactive oxygen/nitrogen species of importance in living organisms include hydroxyl (OH.), superoxide (O2.-), nitric oxide (NO.), nitrogen dioxide (NO2.) and peroxyl (ROO.). Peroxynitrite (OONO-), hypochlorous acid (HOCl), hydrogen peroxide (H2O2), singlet oxygen (1O2), ozone (O3), nitrous acid (HNO2) and dinitrogen trioxide (N2O3) are not free radicals but can easily lead to free radical reactions in living organisms. The term ‘reactive oxygen species’ (ROS) and ‘reactive nitrogen species’ (RNS) is a collective term that includes not only the radicals but also the non-radicals. Oxidative stress is the term referring to the imbalance between generation of reactive oxygen species and the activity of the antioxidant defences.
Humans and other aerobes are able to tolerate oxygen (O2) because, at the same time that organisms were evolving electron-transport chains and other enzyme systems to utilize this molecule, antioxidant defenses to protect against the toxic effects of O2 were evolving in parallel. The aerobic life-style offers great advantages, but is fraught with danger. ROS/RNS are constantly generated in vivo via two main types of processes. ROS/RNS can arise from accidental generation; this encompasses such mechanisms as ‘leakage’ of electrons onto O2 from mitochondrial electron transport chains; nuclear membrane, endoplasmic reticulum (xenobiotic metabolism, prostaglandin synthesis) and hepatocytes (detoxification) contain electron transport systems, cytochrome P-450 and b5, which produce free radicals [6]. Accidental generation also includes the direct reaction of autoxidisable molecules with molecular O2, generating superoxide free radical. The major biological process leading to O2-. generation is the electron transport associated with mitochondrial membrane; ubiquinone-cytochrome b is the most important site of O2- production. It has been estimated that about 1-3% of O2 respired is converted to O2-., a rate that increases during periods of increased energy metabolism. O2- also is produced by phagocytic cells (neutrophils, monocytes, macrophages, eosinophils) and helps them to inactivate viruses and bacteria. When these cells encounter a phagocytable particle, their O2 consumption increases tremendously (‘respiratory burst’) with the activation of a membrane-located enzyme (NADPH-oxidase) which catalyse the reduction of O2 into O2.- . O2.- participate in the production of very reactive chemical species such as OH., hypochlorite and chloramines (Figure 1). The importance of ROS production by the immune system is clearly exemplified by patients with granulomatous disease [7]. These patients have defective membrane-bound NADPH oxidase system thus cannot produce O2-., resulting in multiple and persistent infection, especially Staphylococcus aureus. O2-. is also generated by a variety of cytosolic and membranes-bound enzymes, including xanthine oxidase, cytochrome 450 complex and phospholipase A2. Many biomolecules undergo autooxidation reaction on contact with O2 producing ROS, for example catecholamines, tetrahydrofolates and reduced reduced flavins react directly with O2 to form O2-. [8]. Several sugars, including glucose, react with proteins to produce oxygen radicals. It has been suggested that years of exposure of body tissues to elevated blood glucose in diabetic patients can result in them suffering an ‘oxidative stress’ that may contribute to the side effects of hyperglycaemia [9].
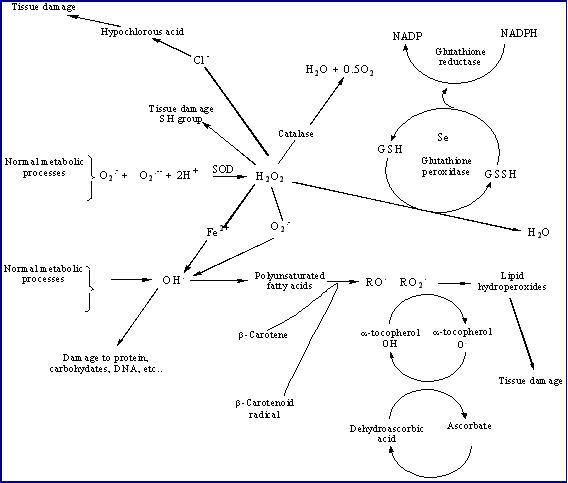 |
Figure 1: Inter-relationship between reactive oxygen species and antioxidants |
H2O2 is a nonradical. It resembles water in its molecular structure and is very diffusible within and between cells. H2O2 is able to diffuse across biological membranes, whereas O2-. does not. As well as arising from dismutation of O2-., H2O2 is produced by the action of several oxidase enzymes in vivo, including amino acid oxidases and the enzyme xanthine oxidases [10]. Xanthine oxidase catalyses the oxidation of hypoxanthine to xanthine, and of xanthine to uric acid; oxygen is simultaneously reduced both to O2-. and to H2O2. Xanthine oxidase is present in many mammalian tissues, especially in the gastrointestinal tract [11]. Phagocytic cells generate substantial amount of H2O2 that is responsible for the cytotoxic action observed in localized tissue inflammation. Peroxisomes contain several ROS generating enzymes including glucose oxidase, amino acid oxidase, xanthine oxidase, glycollate and urate oxidase, as well as flavoprotein oxidases [12]. The major by-product of these oxidases is H2O2, thus explaining the high level of catalase present in peroxisomes which detoxifies H2O2 to H2O. Much of the toxicity of O2.- and H2O2 involves formation of OH. which is the most reactive free radical in vivo with an estimated half-life of about 10-9 sec In the presence of H2O2, superoxide act as the precursor of hydroxyl radical (OH.) (Figure 1). This reaction is very slow and cannot occur in the absence of catalyst; iron ion acts as catalyst for this reaction. The superoxide anion reduces Fe3+ to Fe2+. In the presence of Fe2+, H2O2 readily decompose into OH. and OH-. This reaction is known as the Fenton reaction [13]. Thus, the simultaneous presence of superoxide anion, H2O2, and iron ion lead to the production of hydroxyl radical. Copper ions also react with H2O2 to form OH.. Hydroxyl radical may also be formed by exposure of living organisms to ionising radiation which causes fission of O-H bonds in water, to give H. and OH. [14].
The bioavailability of metal ions is strictly control under normal physiological condition. After absorption from the gut, metal ions are complexed to transport proteins (transferrin, ceruloplasmin). Excess metal ions are stored coupled to storage proteins such as ferritin, haemosiderin. Hydroxyl radical generation can take place when the homeostasis is altered. For example, tissue injury may cause the release of metal ions from damaged cells, contributing to a worsening of the injury. Hydroxyl radical reacts at a diffusion-controlled rate with almost all molecules in living cells. Hence, when hydroxyl radical is formed in vivo, it damages whatever it is generated next to, as hydroxyl radical cannot migrate any significant distance within the cell. OH. attacks proteins, DNA, polyunsaturated fatty acid (PUFA) and many other biological molecules.
HOCl is produced by the neutrophil-derived enzyme myeloperoxidase at sites of inflammation and when activated neutrophils infiltrate reoxygenated tissue [15]. The enzyme oxidizes chloride ions in the presence of hydrogen peroxide (Figure 1). HOCl is not a free radical, but it is a potent chlorinating and oxidizing agent. HOCl reaction with cholesterol causes chlorohydrins that could disrupt cell membranes, leading to cell lysis and death. On the basis of this observation, the cholesterol chlorohydrins have been suggested to be potential biomarkers for oxidative damage associated with neutrophil/monocyte activation [16]. HOCl can attack many other biological molecules. Thiols and thioethers are particularly reactive and other compounds, including ascorbate, urate, pyridine nucleotides and tryptophan are oxidized by HOCl. The main biological chlorination reactions are with amine groups to give chloramines; with tyrosyl residues to give ring chlorinated products; with unsaturated lipids to give chlorohydrins and ring chlorination of cytosine resides in nucleic acids [17].
NO. is formed from the oxidation of L-arginine by nitric oxide synthase (NOS) of which three isoforms are known. NO. has a variety of functions, including memory formation, synaptic plasticity and synaptogenesis. It is thought that the endothelium derived-relaxing factor (EDRF) produced by vascular endothelium, which is an important mediator of vascular responses induced by several pharmacological agents (including brandykinin) is identical to NO. [18, 19]. Excess NO. is cytotxic, both directly (e.g. by combining with tyrosine) and indirectly, by forming ONOO-. Since NO. relaxes smooth muscle in blood vessel walls resulting in lower blood pressure, O2.- by removing NO. can be a vasoconstrictor. Thus, excess vascular O2.- production could contribute to hypertension and vasospasm [20]. ONOO- formed in blood vessel walls may aggravate atherosclerosis by depleting antioxidants and causing peroxidation of LDL. Furthermore, nitration of tyrosine by ONOO- may interfere with cell signal transduction [21]. A role for NO. has also been demonstrated in such human diseases as malaria where NO. appears to be partly involved in resistance to malarial infection, in cardiovascular disease, acute inflammation, cancer, neurodegenerative diseases, and diabetes. Moreover NO. has been implicated in adult respiratory distress syndrome, septic shock, hypertension, thrombosis, renal failure, AIDS encephalopathy, bronchospasm, stroke and male impotence [22].
REACTIVE OXYGEN SPECIES AND THE CARDIOVASCULAR SYSTEM
Reactive oxygen species have been considered deleterious to cell function and there is good evidence to suggest that they play a role in the pathophysiology of cardiac disease states. However, direct cause and effect relationships have not been clearly delineated. The increase in the generation of ROS under several pathophysiological conditions, that seem to be related to inflammatory processes, is still to be comprehensively understood; this may be due to difficulties in defining their site of origin. Impaired mitochondrial reduction of molecular oxygen may be an intracellular source. Secretions by phagocytic white blood cells, dysfunctional endothelial cells, or the auto-oxidation of catecholamines may be the extracellular sources. ROS may also result from cellular injury due to exposure to ionizing radiation, ultraviolet rays, cigarette smoking or other air pollutants. Besides their deleterious effects, ROS are also now being recognized as important regulators of cell function and modulators of cell signalling pathways.
ROS in pathophysiology of Heart disease
One of the strategies used to assess the role of oxidative stress in the pathogenesis of cardiac dysfunction has been to expose isolated cardiac tissues to a defined oxidation stress condition and study the resulting effects [23-31]. Further in vivo and ex vivo studies have provided precious evidence supporting the role of oxidative stress in a number of conditions (atherosclerosis, ischemia-reperfusion injury, hypertension, catecholamine-induced cardiomyopathy, diabetic cardiomyopathy, cardiac hypertrophy and congestive heart failure etc...) leading to severe cardiovascular dysfunctions. In this review the role of ROS in atherosclerosis is being emphasized as, besides being considered as the major cause of morbidity and mortality [32] its outcome is also linked to other conditions leading to cardiovascular disorders. The role of ROS in other above-mentioned conditions has been extensively reviewed and the reader is referred to a number of excellent reports [33, 34].
Most cardiovascular events are secondary to atherosclerosis, a disease of the arteries involving a local thickening of the vessel wall. A stroke or myocardial infarction occurs when the lumen of the vessel becomes completely occluded, usually by a thrombus forming at the site of a plaque. Atherosclerotic lesions are thought to be initiated by emigration of monocytes into the arterial inner core (tunica intima), recruited by adhesion molecules, possibly in response to arterial endothelium injury [35]. A variety of factors have been implicated in causing this initial injury, including mechanical damage from flow stress worsened by high blood pressure, viral infection (herpes viruses and cytomegalovirus), exposure to blood-borne toxins such as xenobiotics from cigarette smoke and elevated levels of normal metabolites, such as glucose, homocysteine or cholesterol [36]. Although a high level of plasma cholesterol is considered to trigger atherosclerosis, the oxidation of cholesterol seems to be a necessary step. In fact, uptake of oxidized low-density lipoprotein (oxLDL) was shown to be an early event leading to the development of atherosclerosis (Figure 2). oxLDL and oxidized lipoproteins have been reported to stimulate O2•- formation leading to apoptosis of cells in the umbilical vascular wall; this was prevented by treatment with antioxidants SOD and catalase [37]. In cultured human coronary artery smooth muscle cells, low levels of oxLDL stimulate the extracellular matrix synthesis indicating the involvement of oxidative stress in the pathogenesis of atherosclerosis [38]. High levels of oxLDL were apoptotic implicating the additive role of ROS in increased plaque vulnerability; this effect was reduced by probucol and catalase [38]. Patients with atherosclerosis and hypercholesterolemia showed higher susceptibility of LDL to oxidation in comparison to patients treated with lipid-lowering agents such as lovastatin and probucol [39].
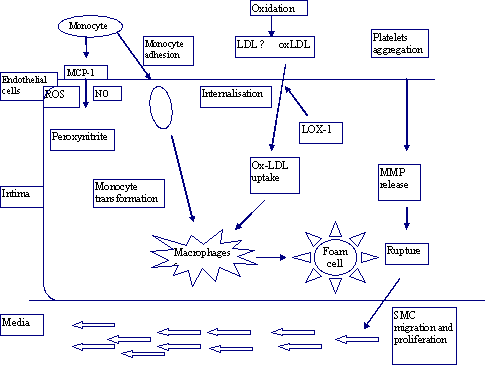 |
Figure 2: ROS and atherosclerosis: increased production of ROS may affect four fundamental mechanisms that contribute to atherosclerosis (i) oxidation of LDL to oxLDL; (ii) endothelial cell dysfunction; (iii) vascular smooth muscle cells migration and proliferation as well as MMPs release; (iv) monocyte adhesion and migration as well as foam cell development due to uptake of ox-LDL. |
In the atherosclerotic lesion produced in the rabbit aorta, significant increases in the iron content were observed suggesting that iron-catalysed free radical reactions may be associated with the development of atherosclerosis [40]. The occurrence of intracellular Ca2+-overload has been proposed as a mechanism of injury due to oxidative stress because human endothelial cells subjected to oxidative stress showed an increase in the level of intracellular Ca2+ and plasma membrane blebbing [41]. Endothelial dysfunction may play an important role in the atherosclerotic process because in patients with atherosclerosis, the antioxidants, probucol and ascorbic acid, improved the endothelium-dependent relaxation suggesting the involvement of ROS in endothelial dysfunction [42]. Increased production of O2•- has been implicated in the impaired endothelium-dependent relaxation in cholesterol fed rabbits and was suggested to be an early event in the hypercholesterolemic atherosclerotic process [43]. Oxidative inactivation of NO• by superoxide has been proposed as a plausible explanation for endothelial dysfunction [44]. When exposed together, O2•- and NO• react with each other three times faster than the reaction rate of O2•- with either Mn2+ and Cu2+/Zn2+-SOD [45]. Therefore, O2•- would preferentially react with NO• rather than SOD and cause inactivation of NOd. In human atherosclerotic arteries, the production of endothelial nitric oxide synthase (the enzyme catalysing NO• formation) as well as NO• has been shown to be depressed [46]. SOD was shown to protect the inactivation of NO• in the canine coronary artery [47]. The generation of O2•- was thought to be due to the activation of the vascular and endothelial enzyme NADH/NADPH oxidase [42]. Moreover, an increase in NADH/NADPH oxidase-dependent vascular O2•- was reported in hypercholesterolaemic rabbits [48]. Oxidation of NO• by O2•- results in the formation of peroxynitrite which could initiate lipid peroxidation or play a role in the oxidation of lipoproteins [49, 50]. Both of the above may be important steps in the development of atherosclerosis.
ROS in Mediated Signal Transduction Pathways in Cardiovascular disorders
The cardiovascular system is a highly complex, well organised system in which signal transduction plays critical physiological and pathophysiological roles (Figure 3). The cellular elements of the heart and vascular wall are equipped with an array of specific receptors and with complex intracellular machinery that facilitates and drives appropriate responses to extracellular stimuli. All vascular cell types, including endothelial cells, smooth muscle cells, adventitial fibroblasts, and resident macrophages, produce ROS [51-55]. Of particular importance in the vasculature are superoxide (O2.-) and hydrogen peroxide (H2O2), since these ROS act as inter- and intra-cellular signaling molecules. The major source of ROS in the vascular wall is non-phagocytic NADPH oxidase, which is regulated by vasoactive agents (Ang II, ET-1, thrombin, serotonin), cytokines (IL-1, TNFa), growth factors (PDGF, IGF-1, VEGF) and mechanical forces (cyclic stretch, laminar and oscillatory shear stress). High levels of low-density lipoprotein (LDL), especially in the form of oxidized low-density lipoprotein (ox-LDL), have also been shown to increase intracellular ROS generation. Under physiological conditions, vascular production of ROS and the consequent activation of redox-dependent signaling pathways and induction of redox-sensitive genes are tightly regulated. However, in pathological conditions, such as in hypertension, atherosclerosis, hyperlipidemia, hyperhomo-cysteinemia, and diabetes, where generation of ROS is increased and the renin angiotensin system may be upregulated, these redox-sensitive events may contribute to cellular processes involved in vascular dysfunction and structural remodeling [56-58].
Redox signalling has been suggested in vascular smooth muscle proliferation, atherosclerosis, angiogenesis, cardiac hypertrophy, fibrosis [59]. Modulation of intracellular signaling pathways (MAPKs), and the subsequent activation of downstream redox sensitive transcription factors like NF-κB, HIF-1, AP-1 results in alterations in gene and protein expression [60] that significantly enhance cardiac dysfunction. Increased bioavailability of vascular ROS leads to VSMC growth, migration, collagen deposition, and altered MMP activity, important factors in arterial remodeling in cardiovascular disease [56-58]. In endothelial cells, oxidative excess induces apoptosis and aniokis (cell shedding), leading to endothelial cell loss and resultant impaired endothelial function. In addition, oxidative stress stimulates activation of transcription factors (e.g., NFkB and AP-1) and pro-inflammatory genes (cytokines, interleukins), upregulation of adhesion molecules (e.g., ICAM, VCAM, PECAM), stimulation of chemokine production (e.g., MCP-1) and recruitment of inflammatory cells (monocytes, macrophages), critical processes involved in vascular inflammation and injury [61]. Increased vascular •O2- and H2O2 also impair endothelium-dependent relaxation, increase contractile reactivity and alter vascular tone. These effects may be mediated directly by elevating cytosolic Ca2+ concentration or indirectly by reducing concentrations of the vasodilator NO•. A common link between free radicals and the aforementioned pathological condition is the disrupted intracellular signal transduction networks and this suggests a rationale for targeting these pathways in chemoprevention.
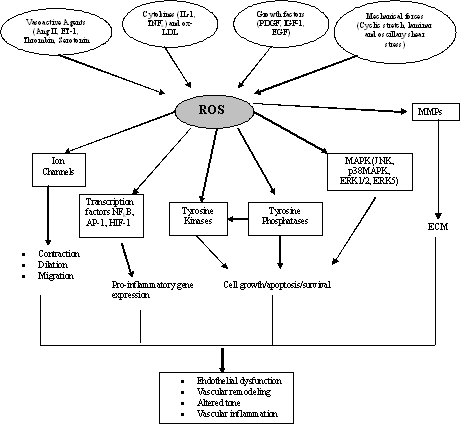 |
Figure 3: Redox-dependent signaling pathways in vascular smooth muscle cells: The increase ROS which is produced from NSDPH oxidase may modify the activity of tyrosine kinases, such as Src, Ras, JAK2, Pyk2, PI3K, and EGFR, as well as mitogen-activated protein kinases (MAPK), particularly p38MAPK, JNK and ERK5. ROS may inhibit protein tyrosine phosphatase activity, further contributing to protein tyrosine kinase activation. ROS also influence gene and protein expression by activating transcription factors, such as NFkB activator protein-1 (AP-1) and hypoxia-inducible factor-1 (HIF-1). ROS stimulate ion channels, such as plasma membrane Ca2+ and K+ channels, leading to changes in cation concentration. Activation of these redox-sensitive pathways results in numerous cellular responses which, if uncontrolled, could contribute to hypertensive vascular damage. |
ANTIOXIDANTS AND THEIR RELEVANCE TO CARDIOVASCULAR DISEASE
An antioxidant has been defined as “any substance that, when present at low concentrations compared with those of an oxidizable substrate, significantly delays or prevents oxidation of that substrate” [38]. When ROS/RNS are generated in vivo, their actions are opposed by intricate and coordinated antioxidant lines of defence systems [61]. These include enzymatic and non-enzymatic antioxidants that keep in check ROS/RNS level and repair oxidative cellular damage (Figure1). The major enzymes, constituting the first line of defence, directly involved in the neutralization of ROS/RNS are: superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPx) (Figure 1) [61]. SOD is a cytoplasmic and mitochondrial enzyme, which accelerate the dismutation of superoxide. There are three forms of SOD: an extracellular and an intracellular copper/zinc (Cu/Zn) and a mitochondrial, manganese (Mn) SOD. All three forms catalyse the dismutation of O2.- to H2O2. Because SOD enzymes generate H2O2, they work in collaboration with H2O2-removing enzymes. CAT, an exclusively peroxisomal enzyme in most tissues, converts H2O2 to water and O2. However, the most important H2O2-removing enzymes are the selenoprotein GPx enzymes. GPx enzymes remove H2O2 by using it to oxidize reduced glutathione (GSH) to oxidized glutathione (GSSG). Glutathione reductase, a flavoprotein enzyme, regenerates GSH from GSSG, with NADPH as a source of reducing power (Figure 3). Glutathione peroxidase also catalyse the reduction of unstable hydroperoxides at the expense of GSH [62].
The second line of defence is represented by radical scavenging antioxidants such as vitamin C, vitamin A and plant phytochemicals like phenolics (emphasised later in this review) that inhibit the oxidation chain initiation and prevent chain propagation [63]. This may also include the termination of a chain by the reaction of two radicals. The repair and de novo enzymes act as the third line of defense by repairing damage and reconstituting membranes. These include lipases, proteases, DNA repair enzymes and transferases [64].
A number of studies have been conducted to explore the roles of various antioxidants in cardiovascular diseases. Diphenylene iodonium (DPI), a potent inhibitor of NADH/NADPH oxidase enzyme has been shown to suppress p38 MAP kinase-mediated VSMC hypertrophy in vitro [65]. Furthermore, it was reported that N-acetyl-L-cysteine (NAC), a radical scavenger and intracellular glutathione precursor, inhibited endothelin-induced ROS generation, JNK activation and VSMC proliferation [66]. Probucol is a modestly potent LDL-lowering agent with powerful anti-oxidant properties that effectively inhibits the oxidative modification of LDL, independent of its lipidlowering effect. By exerting anti-oxidant effect, it may inhibit VCAM-1 and MCP-1 expression and inhibit human aortic SMC proliferation as well as atherogenesis [67].
The HMG-CoA reductase inhibitors, also known as statins, are potent lipid-modifying agents. There is overwhelming evidence from clinical studies that reducing plasma LDL levels with statins, results in a markedly lower risk of cardiovascular events related to atherosclerosis [68]. Recent studies in patients with established CAD show that these agents can cause a modest regression of atherosclerotic lesions. It has been suggested that the antiatherosclerotic effect of statins may be independent of their LDL-lowering effect [69, 70].
AT1R blockers and ACE inhibitors are widely used to treat patients with hypertension and/or congestive heart failure by blocking the effect of Ang II or its formation. Recent studies show that Ang II is also a strong stimulus for ROS generation [71-73] and AT1R blockers as well as ACE inhibitors inhibit the expression of pro-atherogenic factors by decreasing ROS production in vascular endothelial cells and in animal models [74-77]. A large number of studies have provided direct evidence showing the antiatherosclerotic effects of these agents [78-80]. Statins and AT1R blockers exert synergistic effects on the inhibition of atherosclerotic lesions in the apo E-knockout mice placed on a high cholesterol diet [81]. PPAR-γ ligands, which are widely used in the treatment of type II diabetes, have been identified as potent antioxidants [82]. By suppressing NADPH oxidase expression and reducing intracellular ROS production, these agents inhibit the expression of several proatherogenic proteins and apoptosis in vascular endothelial cells and SMCs [83, 84]. Experimental studies show that thiazolidinedione, a potent PPAR-γ ligand, reduces the size and number of atherosclerotic lesions in the vessel wall by modulating foam cell formation and inflammatory responses of macrophages [85].
Vitamins E and C have been demonstrated to reduce the progression of atherosclerosis. Trolox C, a water-soluble vitamin E analogue, and vitamin C (ascorbic acid) abolished the stimulatory effect of Ang II on JNK and p38 activity in VSMC [86]. Intake of vitamin E decreased the incidence of cardiovascular events in the population of ischemic heart disease patients in the Cambridge Heart Antioxidant Study (CHAOS) [87]. Clinical studies show that while these anti-oxidant vitamins do not reduce endpoints related to atherosclerosis, they improve endothelial function by increasing local NO bioavailability and, therefore, endothelium-dependent vasodilation. It has been reported that high dose of vitamin C infusions improved endothelial dysfunction in patients with renovascular hypertension [88]. However, a GISSI-3 study [89] and HOPE study [90] could not show significant beneficial effects of vitamin E in the secondary prevention of coronary artery disease. A Heart Protection Study (HPS) in the UK also could not demonstrate any benefits of vitamin E, vitamin C, and β-carotene combined antioxidants therapy in a large number of high-risk people [91].
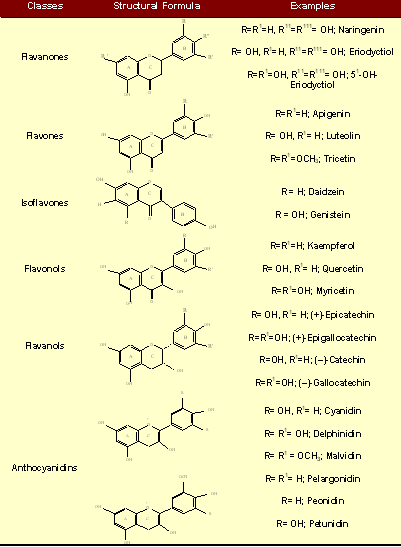
There is increasing interest in phenolics stemming from the context of the "French paradox" [92]. This paradox refers to the correlation of a high-fat and high-cholesterol diet with a lower incidence of coronary heart disease found in Mediterranean cultures and contrasted with a higher incidence of coronary heart disease among most Western cultures. It has been shown that the French paradox may be attributable to regular consumption of red wine and that the unique antiatherogenic effects of red wine reside in the action of polyphenols. Phenolic compounds or polyphenols constitute one of the most numerous and ubiquitously distributed group of plant secondary metabolites, with more than 8000 phenolic structures currently known. Natural polyphenols can range from simple molecules (quinones, phenolic acids,) to highly polymerised compounds (lignins, melanins, tannins), with flavonoids such as flavonols, flavones, isoflavones, flavonones, flavanols and anthocyanins representing the most common and widely distributed sub-group 93 (Table 2).
 |
Phenolics are therefore an integral part of the diet, with significant amounts being reported in vegetables, fruits, teas and traditional plants [94-97]. Although the dietary intake of phenolics varies considerably among geographic regions, it is estimated that daily intake range from about 20 mg to 1 g, which is higher than that for Vitamin E [98]. Epidemiological evidence indicates that consumption of fruit, vegetables and teas may reduce the risk of cardiovascular disease and it is increasingly suggested that this may due to their antioxidants that include ß-carotene, vitamin C, vitamin E and polyphenolics. Dietary antioxidant phenolics may quench reactive oxygen and nitrogen species and, hence potentially modify pathogenic mechanisms relevant to cardiovascular disease. The effectiveness of a dietary antioxidant will depend on a number of factors, such as which ROS or RNS is being scavenged, how and where they are being generated and the accessibility of the antioxidant to possible sites of damage [99]. Many phenolic compounds have been shown to have antioxidant activity in vitro [100] and several observational studies support their role in potentially protecting against cardiovascular disease [101, 102]. However, not all epidemiological studies have found a protective effect of dietary phenolics against heart disease [102, 103]. In vitro studies have used various systems to oxidize LDL and then measure prevention of oxidation after inclusion of phenolic compounds like cinnamic acids [104]. A range of other compounds such as ellagic acid, dimethydiisoeugenol, phenolic rich extracts derived from olive oil, wine, grape, apple and blackcurrent juices have been shown to act as potent antioxidants inhibiting LDL oxidation Ex vivo and/or increasing plasma antioxidant capacity [99] .Tea polyphenols have the inherent capacity to inhibit the development of atherosclerotic lesions by down-regulating genes controlling lipid metabolism, cytokine production and cellular activity within the arterial wall namely genes coding for PPAR-γ, CD36, LXR-α, C-myc coupled with the up-regulation of genes coding for LDL-R and PPAR-α at the transcriptional level [105]. Grape seed proanthocyanidin extract (GSPE), a mixture of 75-80% oligomeric proanthocyanidin and 3-5% monomeric proanthocyanidin has proved its efficacy against the incidence of ischemia-reperfusion injury, apoptosis of cardiomyocytes and reduction of foam cell development. The mechanistic pathways of cardioprotection exerted by the proanthocyanidin rich extract included (1) potent hydroxyl and other free radical scavenging abilities; (2) anti-apoptotic,anti-necrotic and anti-endonucleolytic potentials; (3) modulatory effect on apoptotic regulatory bcl-XL, p53 and c-myc genes; (4) cytochrome P450 2E1 inhibitory activity; (5) inhibitory effects on proapoptotic, cardioregulatory genes c-JUN and JNK-1 and (6) inhibition of vasoconstriction of vascular smooth muscle and endothelium [106, 107].
CONCLUSION
The implication of oxidative stress in the etiology of several chronic and acute degenerative disorders suggests that antioxidant therapy represents a promising avenue for treatment. Strategies for the intervention and prevention of cardiovascular disease require an understanding of the basic molecular mechanism (s) by prophylactic agents (synthetic antioxidants, dietary antioxidant factors from food plants and medicinal plants) that may potentially prevent or reverse the promotion or progression of the disease. It remains unequivocal that emerging scientific support for health claims and identification of active functional ingredients needs to be balanced by addressing toxicological concerns [108]. The real proof of efficacy for existing or novel compounds/extracts should emanate from a demonstration of clinical efficacy on defined therapeutic categories [109]. In this respect, the outcome of one such trial conducted on the Mauritian population on the effects of Mauritian black tea on markers of oxidative stress leading to cardiovascular disease is currently much awaited.
ACKNOWLEDGEMENT
We wish to thank the Mauritius Research Council and the Tertiary Education Commission for their continued interest and support for the research work of the authors. Okezie I Aruoma is Adjunct Research Professor of the University of Mauritius.
LIST OF ABBREVIATIONS
Ang II: Angiotensin II; AP-1: activator protein 1; AT1R: Angiotensin II type 1 Receptor; ECM: extracellular matrix; ERK: extracellular receptor kinase; ET-1: Endothelin-1; HOCl: Hypochlorous acid; HMG-CoA: 3-hydroxy 3-methylglutaryl coenzyme A; ICAM: intercellular adhesion molecule; IGF-1: insulin-like growth factor; IL-1: Interleukin-1; JNK: Jun N-terminal kinase; LDL: low density lipoprotein; MAPK: Mitogen-activated protein kinase; MCP-1: monocyte chemoattractant protein-1; MMPs: matrix metalloproteinases; NF-κB: nuclear factor κB; oxLDL: oxidized low-density lipoprotein; PDGF: platelet-derived growth factor; PECAM: platelet endothelial cell adhesion molecule; PPAR-γ: peroxisome proliferator-activated receptor-γ; Ras: small G-protein; RNS: reactive nitrogen species; ROS: reactive oxygen species; SOD: superoxide dismutase; TNFα: tumor necrosis factor alpha; VCAM: vascular cell adhesion molecule; VEGF: vascular endothelial growth factor; VSMC: vascular smooth muscle cell
REFERENCES
-
Moreno JJ, Mitjavila MT. The degree of unsaturation of dietary fatty acids and the development of atherosclerosis (review). J Nutr Biochem 2003;14:182-95.
-
Central Statistic Office, Mauritius 2005. http://www.gov.mu/portal/site/ncb/cso
-
Moad G, Solomon DH. The Chemistry of FreeRadical Polymerization. Pergamon Press: Oxford,1995.
-
Perkins MJ. Spin trapping. Advances in Physical Organic Chemistry 1980;17:1-64.
-
Bensasson RV, Land EJ, Truscott TG. Excited States and Free Radicals in Biology and Medicine. Contribution from Flash Photolysis and Pulse Radiolysis. Oxford University Press: Oxford 1993.
-
Yu BP. Cellular defenses against damage from reactive oxygen species, Physiol Rev 1994;74:139-62.
-
Curnutte JT, Babior BM. Chronic granulomatous disease. Adv Human Genetics 1987;16:229-45.
-
Halliwell B. Free radicals and antioxidants: A personal view, Nutr Rev 1994;52:253-65.
-
Halliwell B. Drug antioxidant effects: A basis for drug selection. Drugs 1991;42:569-605.
-
Chance B, Sies H, Boveris A. Hydroperoxide metabolism in mammalian organs. Physiol. Rev. 1979;59:527-605.
-
Granger DN. Role of xanthine oxidase and granulocytes in ischemia-reperfusion injury. Am. J. Physiol. 1988;255:1269-75.
-
del Rio LA, Sandalio LM, Palma JM, et al. Metabolism of oxygen radicals in peroxisomes and cellular implications. Free Rad. Biol Med 1992;13:557-80.
-
Koppenol WH. The centennial of the Fenton reaction. Free Rad. Biol Med 1993;15:645-51.
-
Halliwell B. Antioxidants in human health and disease. Annu Rev Nutr 1996;16:33-50.
-
Weiss SJ, Klein R, Slivka A, et al. Chlorination of taurine by human neutrophils. J Clin Invest 1982;70:598-607.
-
Heinecke JW, Li W, Mueller DM, et al. Cholesterol chlorohydrin synthesis by the myeloperoxidase-hydrogen peroxide-chloride system: potential markers for lipoproteins oxidatively damaged by phagocytes. Biochem 1994;33:10127-36.
-
Henderson JP, Byun J, Heinecke JW. Molecular chlorine generated by the myeloperoxidase-hydrogen peroxide-chloride system of phagocyte produces 5-chlorocytosine in bacterial RNA. J. Biol. Chem. 1999;274:33440-8.
-
Palmer RMJ, Ashton DS, Moncada S. Vascular endothelium cell synthesize nitric oxide from L-arginine. Nature 1988;33:664-6.
-
Sneddon JW, Vane JR. endothelium-derived relaxing factor reduces platelet adhesion to bovine endothelium cells. Proc. Natl. Acad. Sci USA 1988;85:1341-4.
-
Nakazono K, Watanabe N, Matsuno, et al. Does superoxide underlay the pathogenesis of hypertension? Proc. Natl Acad Sci 1991;88:10045-8.
-
Darsley-Usmar V, Wiseman H, Halliwell B. Nitric oxide and oxygen radicals: a question of balance. FEBS Lett. 1995;369:131-5.
-
Aruoma OI. Extracts as antioxidant prophylactic agents. Inform 1997;8:1236-42.
-
Kramer JH, Mak IT, Weglicki WB. Differrential sensitivity of canine cardiac sarcolemmal and microsomal enzymes to inhibition by free radical induced lipid peroxidation. Circ Res 1984;55:120-4.
-
Reeves JP , Bailey CA, Hale CC. Redox modification of sodium calcium exchange activity in cardiac sarcolemmal vesicles J. Biol Chem 1986;201:4948-55.
-
Kaneko M., Singal PK, Dhalla NS. Alterations in heart sarcolemmal Ca2+ binding activities due to oxygen radicals. Basic Res Cardiol 1990;85:45-54.
-
Dixon IM, Kaneko M, Hata T, et al. Alterations in cardiac membrane Ca2+ transport during oxidative stress. Mol Cell Biochem 1990;99:125-33.
-
Kaul N, Siveski-Iliskovic N, Hill M, et al. Free radicals and the heart. J. Pharmacol Toxicol Methods 1993;30:55-67.
-
Gupta M, Singal PK. Time course of structure, function and metabolic changes due to an exogenous source of oxygen metabolites in rat heart. Can J Physiol Phramacol 1989;67:1549-59.
-
Mickelson JK, Simpson PJ, Jacson CV, et al. Protection of myocardial function and coronary vasculature by streptokinase J Cardiovasc Phramacol 1988;12:186-95.
-
Kirshenbaum LA, Thomas TP, Randhawa AK, et al. Time course of cardiac myocyte injury due to oxidative stress Mol Cell Biochem 1992;111:25-31.
-
Jolly SR, Kane WJ, Bailie MB, et al. Canine myocardial reperfusion injury: its reduction by the combined administration of superoxide dismutase and catalase. Circ Res 1984;54:277-85.
-
Aviram M. Review of human studies on oxidative damage and antioxidant protection related to cardiovascular diseases Free Rad Res 2000;33:585-97.
-
Griendling KK, Sorescu D, Lassegue, et al. Modulation of protein kinase activity and gene expression by reactive oxygen species and their role in vascular physiology and pathophysiology. Arterioscler. Thromb. Vasc. Biol. 2000;20:2175-83.
-
Dhalla NS, Temsah RM, Netticaden T. Role of oxidative stress in cardiovascular diseases. J. Hypertens. 2000;18:655-73.
-
Steinberg D. Oxidative modification of LDL and atherosclerosis: an update Circulation 1993;95:1062-71.
-
Halliwell B. and Gutteridge J.M.C. 1999 Ch.4, Oxidative stress: adaptation, Damage, repair and Death. In free radicals in Biology and Medicine, 3rd Ed. Oxford University Press, New York.
-
Galle J, Heermeier K, Wanner C. Atherogenic lipoproteins, oxidative stress, and cell death. Kidney Int Suppl1999;71:S62–S65.
-
Bachem MG, Wendelin D, Schneiderhan W, et al. Depending on their concentration oxidized low density lipoproteins stimulate extracellular matrix synthesis or induce apoptosis in human coronary artery smooth muscle cells. Clin Chem Lab Med 1999;37:319–26.
-
Anderson TJ, Meredith IT, Charbonneau F, et al. Endothelium-dependent coronary vasomotion relates to the susceptibility of LDL to oxidation in humans. Circulation1996;93:1647–50.
-
Thong PS, Selley M, Watt F. Elemental changes in atherosclerotic lesions using nuclear microscopy. Cell Mol Biol1996;42:103–10.
-
Geeraerts MD, Ronveaux-Dupal MF, Lemasters JJ, et al. Cytosolic free Ca2+ and proteolysis in lethal oxidative injury in endothelial cells. Am J Physiol1991;261:C889–C896.
-
Anderson TJ, Meredith IT, Yeung AC, et al. The effect of cholesterol-lowering and antioxidant therapy on endothelium-dependent coronary vasomotion. N Engl J Med1995;332:488–93.
-
Ohara Y, Peterson TE, Harrison DG. Hypercholesterolemia increases endothelial superoxide anion production. J Clin Invest 1993;91:2546–51.
-
Harrison DG. Cellular and molecular mechanisms of endothelial cell dysfunction. J Clin Invest1997;100:2153–7.
-
Thomson L, Trujillo M, Telleri R, et al. Kinetics of cytochrome c2+ oxidation by peroxynitrite: implications for superoxide measurements in nitric oxide-producing biological systems. Arch Biochem Biophys1995;319:491–7.
-
Oemar BS, Tschudi MR, Godoy N, et al. Reduced endothelial nitric oxide synthase expression and production in human atherosclerosis. Circulation 1998;97:2494–8.
-
Rubanyi GM, Vanhoutte PM. Superoxide anions and hyperoxia inactivate endothelium-derived relaxing factor. Am J Physiol 1986;250:H822–H827.
-
Warnholtz A, Nickenig G, Schulz E, et al. Increased NADH-oxidase-mediated superoxide production in the early stages of atherosclerosis: evidence for involvement of the renin-angiotensin system. Circulation1999;99:2027–33.
-
Beckman JS, Beckman TW, Chen J, et al. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci USA 1990;87:1620–4.
-
White CR, Brock TA, Chang LY, et al. Superoxide and peroxynitrite in atherosclerosis. Proc Natl Acad Sci USA 1994;91:1044–8.
-
Grech ED, Dodd NJ, Jackson MJ, et al. Evidence for free radical generation after primary percutaneous transluminal coronary angioplasty recanalization in acute myocardial infarction. Am J Cardiol 1996;77:122-7.
-
Slezak J, Tribulova N, Pristacova J. Hydrogen peroxide changes in ischemic and reperfused heart. Cytochemistry and biochemical and X-ray microanalysis. Am J Pathol 1995;147:772-81.
-
Persad S, Panagia V, Dhalla NS. Role of H2O2 in changing beta-adrenoceptor and adenylyl cyclase in ischemia-reperfused hearts. Mol Cell Biochem 1988;86:99-106.
-
Temsah RM, Nitticadan T, Chapman D, et al. Alterations in sarcoplasmic reticulum function and gene expression in ischemic-reperfused rat heart. Am J Physiol 1999;277:H584-H594.
-
Rimm EB, Stampfer MJ, Ascherio A, et al. Vitamin E consumption and the risk of coronary heart disease in men. N Engl J Med 1993;328:1450-6.
-
Berk BC. Redox signals that regulate the vascular response to injury. Thromb Haemostasis 1999;82:810-7.
-
Ferrari R, Ceconi C, Curello S, et al. Oxygen-mediated myocardial damege during ischaemia and reperfusion :role of the cellular defences against oxygen toxicity. J Mol Cell Cardiol 1985;17:937-45.
-
Garlick PB, Davies MJ, Hearse DJ, et al. Direct detection of free radicals in the reperfused rat heart using electron spin resonance spectroscopy. Circ Res 1987;61:757-60.
-
Finkel T. Redox-dependent signal transduction. FEBS Lett 2000;476:52-4.
-
Shah AM, Channon KM. Free radicals and redox signalling in cardiovascular disease. Heart. 2004;90:486-7.
-
Touyz RM, Deschepper C, Park JB,et al. Inhibition of mitogen-activated protein/extracellular signal-regulated kinase improves endothelial function and attenuates Ang II-induced contractility of mesenteric resistance arteries from spontaneously hypertensive rats. J.Hyp 2002;20:1127-1134.
-
Ursini F, Maiorino M, Brigelius-Flohé R, et al. The diversity of glutathione peroxidase. Meth Enzymol. 1995;252:38-63.
-
Willcox JK, Ash AL, Catignani GL. Antioxidants and prevention of chronic disease. Critic Rev Food Sci Nutr 2004;44:275-295.
-
Niki E. Free radicals in chemistry and biochemistry. In Food and Free radicals Ed Hiramatsu et al, Plenum Press, 1997. New York.
-
Ushio-Fukai M, Alexander RW, Akers M, et al. p38 Mitogen-activated protein kinase is a critical component of the redox-sensitive signaling pathways activated by angiotensin II. Role in vascular smooth muscle cell hypertrophy. J Biol Chem 1998;273:15022-9.
-
Fei J, Viedt C, Soto U, et al. Endothelin-1 and smooth muscle cells: induction of jun amino-terminal kinase through an oxygen radical-sensitive mechanism. Arterioscler Thromb Vasc Biol. 2000;273:1244-9.
-
Meng CQ, Somers PK, Rachita CL, et al. Novel phenolic antioxidants as multifunctional inhibitors of inducible VCAM-1 expression for use in atherosclerosis. Bioorg Med Chem Lett 2002;12:2545–8.
-
Semb AG, van Wissen S, Ueland T, et al. Raised serum levels of soluble CD40 ligand in patients with familial hypercholesterolemia: down regulatory effect of statin therapy. J Am Coll Cardiol 2003;41:275–9.
-
Tsiara S, Elisaf M, Mikhailidis DP. Early vascular benefits of statin therapy. Curr Med Res Opin 2003;19:540–56.
-
Bonetti PO, Lerman LO, Napoli C, et al. Statin effects beyond lipid lowering—are they clinically relevant? Eur Heart J 2003;24:225–248.
-
Lo YY, Wong JM, Cruz TF. Reactive oxygen species mediate cytokine activation of c-Jun NH2-terminal kinases. J Biol Chem 1996;271:15703–7.
-
Watanabe Y, Suzuki O, Haruyama T, et al. Interferon-gamma induces reactive oxygen species and endoplasmic reticulum stress at the hepatic apoptosis. J Cell Biochem 2003;89:244–53.
-
Cheng TH, Cheng PY, Shih NL, et al. Involvement of reactive oxygen species in angiotensin II-induced endothelin-1 gene expression in rat cardiac fibroblasts. J Am Coll Cardiol 2003;42:1845–54.
-
Singh BM, Mehta JL. Interactions between the renin-angiotensin system and dyslipidemia: relevance in the therapy of hypertension and coronary heart disease. Arch Intern Med 2003;163:1296–1304.
-
Chen H, Li D, Sawamura T, et al. Upregulation of LOX-1 expression in aorta of hypercholesterolemic rabbits: modulation by losartan. Biochem Biophys Res Commun 2000;276:1100–1104
-
Mehta JL, Li DY, Yang H, et al. Angiotensin II and IV stimulate expression and release of plasminogen activator inhibitor-1 i cultured human coronary artery endothelial cells. J Cardiovasc Pharmacol 2002;39:789–94.
-
Li D, Chen H, Mehta JL. Angiotensin II via activation of type 1 receptor upregulates expression of endoglin in human coronary artery endothelial cells. Hypertension 2001;38:1062–7.
-
Takai S, Kim S, Sakonjo H, et al. Mechanisms of angiotensin II type 1 receptor blocker for anti-atherosclerotic effect in monkeys fed a high-cholesterol diet. J Hypertens 2003;21:361-9.
-
Nickenig G, Harrison DG. The AT(1)-type angiotensin receptor in oxidative stress and atherogenesis: Part II: AT(1) receptor regulation. Circulation 2002;105:530–6.
-
Hayek T, Attias J, Coleman R, et al. The angiotensin-converting enzyme inhibitor, fosinopril, and the angiotensin II receptor antagonist, losartan, inhibit LDL oxidation and attenuate atherosclerosis independent of lowering blood pressure in apolipoprotein E deficient mice. Cardiovasc Res 1999;44:579–87.
-
Chen J, Li D, Schaefer R, et al. Cross-talk between dyslipidemia and renin-angiotensin system and the role of LOX-1 and MAPK in atherogenesis studies with the combined use of rosuvastatin and candesartan. Atherosclerosis 2006;184:295-301.
-
Inoue I, Goto S, Matsunaga T, et al. The ligands/activators for peroxisome proliferator-activated receptoralpha (PPAR alpha) and PPAR gamma increase Cu2+, Zn2+- superoxide dismutase and decrease p22phox message expressions in primary endothelial cells. Metabolism 2001;50:3-11.
-
Braunstein S. Cardiovascular disease and benefits of thiazolidinediones. Postgrad Med 2003;99:45–52.
-
Varo N, Vicent D, Libby P, et al. Elevated plasma levels of the atherogenic mediator soluble CD40 ligand in diabetic patients: a novel target of thiazolidinediones. Circulation 2003;107:2664–9.
-
Wang M, Tafuri S. Modulation of PPAR gamma activity with pharmaceutical agents: treatment of insulin resistance and atherosclerosis. J Cell Biochem 2003;89:38–47.
-
Kyaw M, Yoshizumi M, Tsuchiya K, et al. Antioxidants inhibit JNK and p38 MAPK activation but not ERK 1/2 activation by angiotensin II in rat aortic smooth muscle cells. Hypertens Res 2001;24:251-61.
-
Stephens NG, Parsons A, Schofield PM, et al.Randomised controlled trial of vitamin E in patients with coronary disease: Cambridge Heart Antioxidant Study (CHAOS). Lancet 1996;347:781-6.
-
Higashi Y, Sasaki S, Nakagawa K, et al. Endothelial function and oxidative stress in renovascular hypertension. N Engl J Med 2002;346:1954-62.
-
GISSI-Prevenzione Investigators. Dietary supplementation with n-3 polyunsaturated fatty acids and vitamin E after myocardial infarction: results of the GISSI-Prevenzione trial. Gruppo Italiano per lo Studio della Sopravvivenza nell'Infarto miocardico. Lancet 1999;354:447-55.
-
Yusuf S, Dagenais G, Pogue J, et al. Vitamin E supplementation and cardiovascular events in high-risk patients. The Heart Outcomes Prevention Evaluation Study Investigators. N Engl J Med 2000;342:154-60.
-
MRC/BHF Heart Protection Study of antioxidant vitamin supplementation in 20 536 high-risk individuals: a randomised placebo-controlled trial. Lancet 2002;360:23-33.
-
Renaud S, de Lorgeril M. Wine, alcohol, platelets and the French Paradox for coronary heart disease. Lancet 1992;339:1523-6.
-
Bravo L. Polyphenols: chemistry, dietary sources, metabolism, and nutritional significance. Nutr Rev. 1998;56:317-33.
-
Bahorun T, Luximon-Ramma A, Crozier A, et al. Total phenol, flavonoid, proanthocyanidin and vitamin C levels and antioxidant activities of Mauritian vegetables, J Sci Food Agric. 2004;84:1553-61.
-
Luximon-Ramma A, Bahorun T, Crozier A. Antioxidant actions and phenolic and vitamin C contents of common Mauritan exotic fruits. J Sci Food Agric. 2003;83:496-502.
-
Luximon-Ramma A, Bahorun T, Crozier A, et al. Characterization of the antioxidant functions of flavonoids and proanthocyanidins in Mauritian black teas. Food Res Int. 2005;38:357-67.
-
Luximon-Ramma A, Bahorun T, Crozier A, et al. Assessment of the polyphenolic composition of the organic extracts of Mauritian black teas: A potential contributor to the antioxidant function of the endemic teas Biofactors, In press.
-
Hollman PC, Katan MB. Bioavailability and health effects of dietary flavonols in man. Arch Toxicol Suppl. 1998;20:237-48.
-
Morton LW, Caccetta RA, Puddey IB, et al. Chemistry and biological effects of dietary phenolic compounds: relevance to cardiovascular disease. Clin Exp Pharmacol and Physiol 2000;27:152-9.
-
Shahidi F, Wanasundara PKJPD. Phenolic antioxidants. Crit. Rev. Food. Sci. Nutr. 1992;32:67-103.
-
Renaud S, Lorgeril M. The French paradox: Dietary factors and cigarette smoking-related health risks. Ann N.Y. Acad. Sci 1993;686:299-309.
-
Hertog MGL, Sweetman PM, Fehily AM, et al. Antioxidant flavonols and ischemic heart disease in a Welsh population of men: The Caerphilly Study. Am. J. Clin. Nutr. 1997;65:1489-94.
-
Rimm EB, Katan MB, Ascherio A, et al. Relation between intake of flavonoids and risk of coronary heart disease in male health professionals. Ann Intern. Med. 1996;125:384-9.
-
Nardini M, D’Aquino M, Tomassi G, et al. Inhibition of human low-density lipoprotein oxidation by caffeic acid and other hydroxycinnamic acid derivatives Free Rad. Biol Med 1995;19:541-52.
-
D Sikand K, Shukla AR. Effect of green tea polyphenols on the genes with atherosclerotic potential. Phytother Res 2004;18:177-9.
-
Bagchi D, Sen CK, Ray SD, et al. Molecular mechanisms of cardioprotection by a novel grape seed proanthocyanidin extract. Mutat Res. 2003;9462:1-113.
-
Pernow J, Wang QD. Endothelin in myocardial ischaemia and reperfusion. Cardiovasc Res. 1997;33:518–26.
-
Aruoma OI, Bahorun T, Clement Y, et al. Inflammation, Cellular and redox signaling mechanisms in cancer and degenerative diseases. Mut. Res. 2005;579:1-5.
-
Aruoma O.I., Sun B., Fujii S, et al. "Low molecular proanthocyanidin dietary biofactor Oligonol®: Its modulation of oxidative stress, bioefficacy, neuroprotection, food application and chemoprevention potentials" Biofactors (in press).