INTRODUCTION
Cardiovascular disease (CVD) continues to be the leading cause of death for both men and women, accounting for approximately 40% of all annual deaths. Among the various cardiovascular diseases, coronary artery disease is the single largest killer. World Health Organization (WHO) estimates that 17 million people die of cardiovascular disease annually, of which 7.2 million deaths are attributable to coronary heart disease [1]. In some East, Central, and Southern Africa (ECSA) countries, such as Mauritius and Seychelles, CVD is the leading cause of mortality, responsible for more than one-third of deaths [2]. In Mauritius Death statistics over the past few years show that over 80% of deaths are due to non communicable diseases. In 2001, 40% of deaths in people of working age were from diseases of the circulatory system, including hypertension however in 2005 approximately 51% of deaths were attributed to cardiovascular diseases [3]. Cardiovascular disease is a combination of multifactorial risk factors including hypercholesterolemia, hypertension, smoking, diabetes, sedentary life style, consumption of high fat diet, family history of early MI and stress. This may be the result of acute ischemia-reperfusion injury, endothelial damage of hyperhomocysteinemia, as well as chronic oxidative damage secondary to lipid peroxidation [4].
Over recent decades, a large body of evidence has accumulated indicating that free radicals play a critical role in cellular processes implicated in atherosclerosis. Free radicals are any reactive organic or inorganic molecules with one or more unpaired electrons, commonly formed in the body as a result of metabolic processes which are normally eliminated by the antioxidant line of defense system of the body [5].
However the free radicals or reactive oxygen species (ROS) that are not destroyed can be toxic because of their propensity to react with biological molecules such as lipids, proteins and DNA may cause oxidative-stress induced damage. Excessive production of ROS, outstripping endogenous antioxidant defense mechanisms, is referred to as oxidative stress [6]. Accumulated evidence has shown that reactive oxygen species are also the important mediators of cell signaling events such as inflammatory reactions (superoxides) and the maintenance of vascular tone (nitric oxide). However the overproduction of ROS such as superoxide has been associated with the pathogenesis of variety of diseases including cardiovascular diseases, diabetes, cancers, Alzheimer’s disease, retinal degeneration, ischemic dementia and other neurodegenerative disorders and ageing [7-9].
Large numbers of studies have been conducted so far, showing the contributive role of quercetin to prevent cardiovascular diseases [10-14]. This article will review the process of oxidative stress mediated atherogenesis and their signaling pathway through which they relate to the cardiovascular diseases. The role of various antioxidative therapies with more emphasis on “Quercetin”- a flavonoid will be discussed.
OXIDATIVE STRESS AND ATHEROGENESIS
Figure 1 demonstrates various steps where oxidative stress could be involved in atherogenesis [15]. In general, increased production of ROS may affect four fundamental mechanisms that contribute to atherosclerosis: endothelial cell dysfunction, vascular smooth muscle cells (VSMC) growth, monocyte migration and oxidation of LDLs (ox LDL) [16, 17].
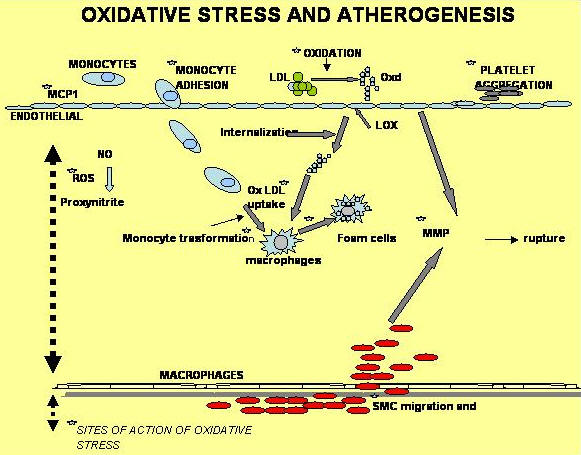 |
Endothelial cell dysfunction
Risk factors Including hyperlipidemia, hypertension, diabetes, smoking, all are associated with the overproduction of ROS or increase oxidative stress. They are considered to be major factors pf the pathogenesis of endothelial dysfunctions and markers of unfavorable cardiovascular prognosis [18].
Endothelial dysfunction is associated with an increased risk of cardiovascular events. There have been at least eight studies involving over 1500 subjects and all indicate that endothelial dysfunction has prognostic value [19-26]. Endothelial dysfunction is characterized by reduction of the bioavailability of vasodilators, in particular nitric oxide (NO), whereas endothelin-derived contracting factors are increased [27]. This imbalance leads to the impairment of endothelium–dependant vasodilatation which represents the functional characteristics of endothelial dysfunctions. On the other hand, endothelial dysfunctions aside from denoting impaired endothelium dependant vasodilatation also comprises a specific state of endothelial activation, which is characterized by a proinflammatory, proliferative and procoagulatory milieu that favors all the stages of atherogenesis [28].
Vascular smooth muscle cells (VSMC) growth
Proliferation of vascular smooth muscles is a characteristic feature of atherosclerosis and ROS can induce vascular SMC growth. The increase in SMC growth by ROS occurs as a result of stimulation of the expression of fibroblast growth factors (FGF) and fibroblast growth factor receptor-1 FGFR-1), Insulin like growth factor-1(IGF-1), Insulin like growth factor-1 receptor (IGF-1R) as well as epidermal growth factor receptor (EGFR) [29-32]. Also there is increasing evidence that ROS generation, via NADPH oxidase activation plays a critical role in Ang II-induced vascular SMC proliferation and hypertrophy [33]. Several studies showed that very large amount of ROS may even induce vascular SMC death by either apoptosis or necrosis [34].
Monocyte migration
Increase adhesion of monocytes to endothelial cells has also been linked to the development and progression of atherosclerosis in humans. One study has shown that the effect of increased concentration of glucose (as in diabetes; important risk factor of CVD) on monocyte adhesion to endothelial cells is due to increased production of ROS [35]. A number of studies have shown that ROS unregulated the expression of intracellular adhesion molecules-1 (ICAM-1) [31], vascular cell adhesion molecule-1(VCAM-1) [36, 37], monocyte chemoattractant protein-1 (MCP-1) [38], P-selectin [39], L-selectin [40], E-selectin [35] and platelet endothelial cell adhesion molecule-1 (PECAM-1) [41] in vascular endothelial cells.
Oxidation of LDLs (ox- LDL)
A large body of literature has confirmed a central proatherogenic role of ox-LDL in vascular cells [42-45]. Increasing evidence has shown that ox-LDL is not only a marker of oxidative stress, but itself can induce oxidative stress in vascular tissues [46, 47]. It has been recently shown that ox-LDL induces proatherosclerotic NADPH oxidase expression and superoxide anion formation in human vascular endothelial cells [48], and this may be one of the mechanism by which ox-LDL stimulates ROS generation and the resultant endothelial dysfunctions as well as atherosclerosis. Strong evidence in favor of pro-atherosclerotic role for ox-LDL comes from a number of studies demonstrating the noxious effects of ox-LDL on various components of arterial wall. For example ox-LDL causes activation of endothelial cell lining the arterial wall, resulting in the expression of several adhesion molecules that facilitate the adhesion of monocytes/macrophages [42]. Ox-LDL also activates inflammatory cells and facilitates the release of number of growth factors from monocytes/macrophages [43, 44]. Vascular SMCs exhibit intense proliferation when exposed to ox-LDL [42] and also enhances the formation of matrix metalloproteinase (MMPs) in vascular endothelial cells and fibroblasts, thus setting the stage wherein oxidative stress leads to rupture of a soft plaque [45]. In addition ox-LDL up regulates the expression of endothelial receptors LOX-1 and other scavenger receptors mainly expressed on macrophage/monocyte. The increased expression of these receptors is responsible for the uptake of ox-LDL and the formation of foam cells, which is an early step in atherogenesis. Angiotensin II (Ang II) enhances the uptake of ox-LDL and biosynthesis of cholesterol in macrophages, leading to the formation of foam cells [49, 50]. Angiotensin II up regulates LOX-1 gene and protein expression in cultured human coronary artery endothelial cells and enhances the noxious effects of ox-LDL [49].
OXIDATIVE- STRESS MEDIATED SIGNALING PATHWAY
Although the observations discussed above implicate oxidative-stress induced ROS as a stimulus for the expression and release of several pro-atherogenic and inflammatory mediators, little is known regarding the specific intracellular signaling pathways by which ROS act [50]. It is increasingly suggested that signaling in response to pro-atherogenic agents requires ROS [51]. Pro-atherogenic agents comprise a large variety of molecules. For examples cytokines, including tumor necrosis factor-γ (TNF-γ), interferon-γ (IFN-γ), interleukin-1,-6 (IL-1, IL-6), and angiotensin II (Ang II), [52–54] stimulate intracellular generation of ROS. High levels of low-density lipoprotein (LDL), especially in the form of oxidized low-density lipoprotein (ox-LDL), have also been shown to increase intracellular ROS generation [55]. In addition, growth factors, such as platelet-derived growth factor (PDGF) and epidermal growth factor (EGF) as well as vascular endothelial growth factor (VEGF), and hormones, such as insulin, all greatly induce intracellular ROS generation [56, 57].
Some recent studies suggest that the function and activity of intracellular signals are regulated by ROS. These studies link the cellular oxidative state to specific mitogen-activated protein kinases (MAPKs), such as p38 kinase [49] and to extracellular signal-regulated kinase 1/2 (ERK 1/2), [56] as well as the cJun N-terminal kinase (JNK) [57] signaling pathway. MAP kinase activation regulates cell differentiation, growth and apoptosis through phosphorylation of downstream target proteins and activation of transcription factors. Ang II and endothelin-1 are other activators of MAP kinases in VSMC and are believed to cause vascular remodeling [58, 59]. Interestingly, Ang II potently activates ERK1/2 and p38; however, only p38 is sensitive to both inhibition of NADH/NADPH oxidase activity and catalase overexpression in VSMC [60, 61]. Vasoactive peptide endothelin-1 stimulated proliferation of human coronary artery smooth muscle cells occurs through the activation of ERK1/2 and transcription factor activator protein-1 (AP-1) [59]. It was also reported that MAP kinase mediated the increase in LOX-1 expression in endothelial cells [62].
Although MAPK plays a central role in ROS-mediated signal transduction in cardiovascular diseases, other protein kinases are also involved in ROS-mediated changes in vascular cells [63]. Protein tyrosine kinases are among the intracellular mediators that may be rapidly activated by many stimuli including ROS. Several tyrosine kinases are activated by ROS, and the Src family of kinases has been most frequently described [63].
Phospholipases may also be activated by ROS [63]. At least three important phospholipases have been shown to be activated by ROS in vascular cells, including phospholipase A2, [64] phospholipase C [65] and phospholipase D [66]. Phospholipase A2 is similarly activated by increase in intracellular calcium caused by ROS. It is important to note that many lipids generated by phospholipase A2 may themselves generate ROS through the action of monooxygenases. Phospholipase C is a calcium dependent phospholipase that hydrolyzes PIP2 to generate IP3 and diacylglycerol. Although there are no published findings to suggest that ROS directly activate phospholipase C, generation of ROS may be an early event in growth factor-dependent signaling and activation of phospholipase C. Phospholipase D has been shown to be stimulated by H2O2, fatty acid hydroperoxides, and 4-hydroxynonenal in endothelial cells. Further studies are required to elucidate the mechanisms by which phospholipases are activated [63].
In addition to regulating enzyme activity, ROS participate in signal transduction by generating classic second-messengers (calcium and lipid mediators) that transmit signals to intracellular mediators in both the cytoplasm and nucleus [63]. Other important mediators of the ROS-sensitive signaling pathway are small G proteins. Based on experiments with recombinant p21 Ras in vitro, it was shown that ROS directly promote guanine nucleotide exchange on p21 Ras [67]. This leads to an increased population of active p21 Ras-GTP, resulting in increased availability of p21 Ras to its downstream targets [68].
The final step in the ROS-mediated signaling pathway usually involves the activation of transcription factors, which are proteins that are transported to the nucleus upon activation and trigger target gene expression. Nuclear factor-κB (NF-κB) is an inducible transcription factor that is a likely target for ROS signal transduction. The recognition sequence for NF-κB is present in genes, such as E-selectin, VCAM-1 and ICAM-1, whose expression causes monocyte adhesion to endothelial cells. [69]
In addition to NF-κB, activator protein-1 (AP-1) also appears to be activated by ROS. Regulation of AP-1 by ROS has been shown to involve p38 MAPK and ERK1/2 as well as JNK in vascular SMCs [70]. Other transcription factors such as hypoxia-inducible factor-1 (HIF-1) and early growth response-1 (Egr-1) may also be involved in ROS-mediated pro-atherogenic events in vascular SMCs and endothelial cells [71, 72]. Figure 2 outlines the ox-LDL and Ang II-induced ROS signaling pathways [15].
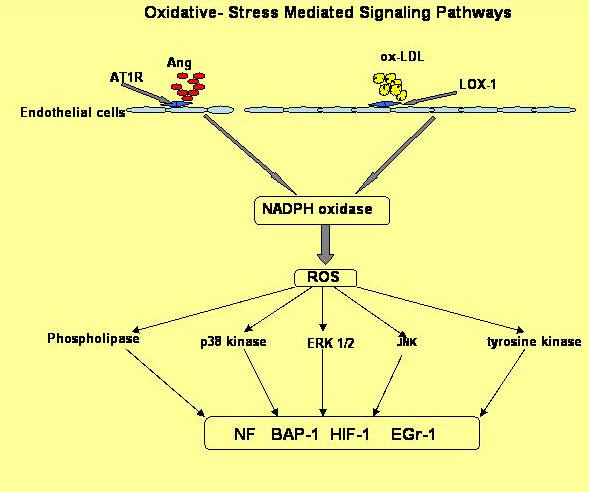 |
ANTIOXIDATIVE THERAPIES
Given that oxidative stress plays a pivotal role in the pathogenesis of cardiovascular diseases, it is not surprising that antioxidant therapies are one of the most effective and promising strategies against atherogenesis. To date, there are at least five groups of anti-atherosclerotic agents that have exhibited anti-oxidant effects, albeit to varying degrees [73] (1) probucol; (2) HMG-CoA reductase inhibitors; (3) AT1R blockers and ACE inhibitors; (4) peroxisome proliferator-activated receptor-γ (PPAR-γ) ligands; (5) vitamins C, E and Bioflavonoids.
Probucol is a modestly potent LDL-lowering agent with powerful anti-oxidant properties that effectively inhibits the oxidative modification of LDL independent of its lipid lowering effect and also inhibits VCAM-1 and MCP-1 expression and inhibits human aortic SMC proliferation as well as atherogenesis [74].
HMG-CoA reductase inhibitors, also known as statins, are potent lipid-modifying agents. There is overwhelming evidence from clinical studies that reducing plasma LDL levels with statins results in a markedly lower risk of cardiovascular events related to atherosclerosis [75]. Recent studies in patients with established CAD show that these agents can cause a modest regression of atherosclerotic lesions. It has been suggested that the anti-atherosclerotic effect of statins may be independent of their LDL-lowering effect, among which is their ability to decrease ROS generation. [76, 77]
AT1R blockers and ACE inhibitors are widely used to treat patients with hypertension and/or congestive heart failure by blocking the effect of Ang II or its formation. Recent studies show that Ang II is also a strong stimulus for ROS generation and AT1R blockers as well as ACE inhibitors inhibit the expression of pro-atherogenic factors by decreasing ROS production in vascular endothelial cells and in animal models. [78, 79]
Peroxisome proliferator-activated receptor-γ (PPAR-γ) ligands, a member of the nuclear receptor super-family, were initially developed to affect glucose and lipid metabolism. These agents, such as rosiglitazone and pioglitazone, are widely used in the treatment of type II diabetes. Recently, PPAR-γ ligands have been identified as potent anti-oxidants. [80] By suppressing NADPH oxidase expression and reducing intracellular ROS production, these agents inhibit the expression of several proatherogenic proteins and apoptosis in vascular endothelial cells and SMCs [81, 82]. Experimental studies show that thiazolidinedione, a potent PPAR-γ ligand, reduces the size and number of atherosclerotic lesions in the vessel wall by modulating foam cell formation and inflammatory responses of macrophages [83]. A recent study [84] shows a significant vascular protective effect of PPAR-γ ligand rosiglitazone in hypercholesterolemic rabbits, most likely due to the attenuation of oxidative stress. This endothelial protective effect of rosiglitazone may reduce leukocyte accumulation in vascular walls and contribute to its antiatherosclerotic effect. The other ligand, pioglitazone, decreases TNF-α-mediated apoptosis in human coronary artery endothelial cells by reducing ROS formation [85]. Thus, PPAR-γ ligands have the potential for the treatment of atherosclerosis and type II diabete [86].
Vitamins E and C have been demonstrated to reduce the progression of atherosclerosis in animal models [87, 88] but this effect is not consistently observed. Clinical studies show that while these anti-oxidant vitamins do not reduce endpoints related to atherosclerosis, they improve endothelial function by increasing local NO bioavailability and, therefore, endothelium-dependent vasodilation. [89]
Bioflavonoids, which are polyphenolic compounds, are believed to be beneficial for the prevention and treatment of atherosclerosis and cardiovascular diseases. Recently, much interest in flavonoids has been generated from the findings of the French paradox [90]. This paradox refers to the correlation of a high-fat and high-cholesterol diet with a lower incidence of coronary heart disease found in Mediterranean cultures. It has been shown that the French paradox may be attributable to regular consumption of red wine and that the unique anti-atherogenic effects of red wine reside in the action of polyphenols. Alongside the French paradox, the REGICOR Study has shown another paradox in the Mediterranean area: a high prevalence of cardiovascular risk factors with low incidence of myocardial infarction in the population of Girona, Spain. The antioxidant effects associated with olive oil consumption could explain part of this Mediterranean Paradox [91, 92]. Quercetin-derived compound hydroxytyrosol possesses high antioxidant property, decreases the susceptibility of LDL to oxidation [93-95].
Many others epidemiological studies also suggest that flavonoid-rich diets high in fruits and/or vegetables reduce the risk of coronary heart disease [96-98]. Based on a review of more than 250 observational studies fruits and vegetables were reported to be important source of flavonoids, beneficial in the prevention of cancer and cardiovascular disease [99]. Cardiovascular deaths were reduced by 16% (8,000 deaths annually) by increasing the current 250 g of fruits and vegetables per day to 400 g per day in the Dutch general population [99].
In the Zutphen Elderly Study, researchers evaluated the diets of 805 Danish men between the ages of 65 and 84. The subjects were then followed up for five years. It was found that the men consuming highest flavonoid levels were slightly less than half as likely to have suffered a heart attack during the course of the study [100]. A recent meta-analysis of seven prospective cohort studies with 105,000 individuals indicated that high dietary intake of flavonoids from a small number of fruits and vegetables, tea and red wine are inversely associated with coronary heart disease risk [101].
Bioflavonoids occur ubiquitously in foods of plant origin. Over 4000 different flavonoids have been described, and they are categorized into flavonols, flavones, catechins, flavanones, anthocyanidins, and isoflavonoids [102].
Quercetin 3, 3´, 4´, 5, 7-Penta-hydroxyflavon is one of the most widely distributed bioflavonoids (flavonol), which are abundant in red wine, tea, and onions. This is the most abundant flavonol in the diet which possesses biological activities such as antioxidative [103, 104], anticarcinogenic [105, 106] and enzyme-inhibiting activities [107-109]. However numerous other in vitro studies have revealed diverse biological effects of quercetin, including apoptosis induction, antimutagenesis, protein kinase C (PKC) inhibition, lipoxygenase inhibition, histamine-release inhibition, superoxide dismutase (SOD)-like activity, modulation of cell cycles, angiogenesis inhibition, and inhibition of angiotensin converting enzyme [110].
It is noteworthy here that quercetin exists rarely as aglycon in foods and plants. It mainly occurs in glycosylated forms and the associated sugar moiety is very often either glucose or rhamnose. However other sugars may also be involved like galactose, arabinose, xylose, or glucuronic acid. Quercetin-3-rutinoside and quercetin-4'-glucoside are the important forms of quercetin in food. Quercetin-3-rutinoside accounts for ~40% of quercetin in black tea [111], however quercetin-4'-glucoside accounts for ~45% of quercetin in onions [112]. More importantly it is the sugar moiety in quercetin glycosides which maximally affects the bioavailability in humans like the bioavailability of quercetin-3-rutinoside is only 20% of that of quercetin-4'-glucoside [113]. The bioavailability of quercetin-3-glucoside is similar to that of quercetin-4'-glucoside but the glucoside form is more readily absorbed than glycoside forms [113, 114]. The mechanism for quercetin absorption is not known. However Hollman et al [113, 115] speculated that the intestinal sodium-glucose co-transporter is able to transport glucose attached to quercetin through the intestinal cell. With regard to bioactivity of various forms, quercetin conjugated with glycosides, glucuronic acid or sulfates also has antioxidant activity in vitro, although the antioxidant activity is lower than that of the quercetin aglycone [116, 117]. Fifty percent of ingested quercetin glucosides, is absorbed in the small intestine and subsequently metabolized into isorhamnetin, in the liver and in other organs. The 50% of ingested quercetin which is not absorbed in the small intestine is metabolized by the colonic microflora into quercetin aglycone and phenolic acids which might be absorbed from the colon [116, 117]. Only 3% of the ingested quercetin is recovered in urine as aglycone or its conjugates. Metabolites of quercetin may also be biologically important, because they also have antioxidant activity in vitro [117] and might exert antioxidant effects in humans.
QUERCETIN ACTION ON CARDIOVASCULAR DISEASES
Antioxidative action
Oxidized low density lipoproteins are atherogenic, and are considered to be a crucial intermediate in the formation of atherosclerotic plaques. Oxidized low density lipoproteins cholesterol (LDL cholesterol) is taken up more readily by macrophages, which leads to the formation of foam cells and atherosclerotic plaque [118]. Oxidised LDL has several properties that promote atherogenesis, apart from its rapid uptake into macrophages via the scavenger receptor. An oxidised form of LDL is chemotactic for circulating macrophages and smooth muscle cells and facilitate monocyte adhesion to the endothelium and entry into the subendothelial space and inhibits the release of nitric oxide and the resulting endothelium dependent vasodilation [119, 120]. Oxidised LDL can induce arterial wall cells to produce chemotactic factors, adhesion molecules, cytokines, and growth factors that have a role to play in the development of the plaque [121, 122]. Mechanisms that prevent or slow down this chain of events may decrease the risk of coronary heart diseases. The capacity of flavonoids to act as antioxidants depends upon their molecular structure. The position of hydroxyl groups and other features in the chemical structure of flavonoids are important for their antioxidant and free radical scavenging activities. Among all the flavonoids quercetin is the most potent antioxidant whose high reactivity could be associated with the presence of the OH group in B- and the C- ring [123]. Sim et al investigated the structure-activity relationship of flavonoid compounds on their antioxidant property and inhibitory effects against the matrix metalloproteinases (MMP) activity. The effects of several flavonoids; myricetin, quercetin, kaempferol, luteolin, apigenin and chrysin, on the reactive oxygen species scavenging activity and inhibitory effect against the MMP activity were examined.The antioxidant property of quercetin was found to be more than Kaempferol which was correlated with the respective number of OH group on their B-ring [124].
Infact Silva et al [125] has shown that quercetin is an even stronger antioxidant than ascorbic acid, alpha-tocopherol, and flavones. However in another study the antioxidant abilities of several polyphenols were determined, and quercetin was most active among all [126].
Other studies have also demonstrated that quercetin inhibits lipid peroxidation effectively by scavenging free radicals and/or chelating transition metal ions [127] including superoxide anions [128] singlet oxygen [129] and lipid peroxy-radicals [130]. Quercetin inhibits the iron ion-dependent lipid peroxidation systems by chelating iron ions with the formation of inert iron complexes. Quercetin is able to suppress free radical processes at three stages: the formation of superoxide ion, the generation of hydroxyl radicals in the Fenton reaction and the formation of lipid peroxy radicals [131].
Antiplatelets action
Platelets are involved in atherosclerotic disease development and the reduction of platelet activity by medication reduces the incidence and severity of the disease. Platelet aggregation contributes to both the development of atherosclerosis and acute platelet thrombus formation Activated platelets adhering to vascular endothelium generate lipid peroxides and oxygen free radicals, which inhibit the endothelial formation of prostacyclin and nitrous oxide. However quercetin directly scavenges free radicals, thereby maintaining proper concentrations of endothelial prostacyclin and nitric oxide [132]. An experimental study in dogs and monkeys showed quercetin to be the most effective inhibitor of platelet aggregation. [133]
In one of the randomized cross-over trials in human designs, a comparative study was done with ten healthy human subjects to evaluate the efficacy of purple grape fruit juice, orange juice or grapefruit on the platelet aggregation. The purple grape juice, which is rich in quercetin content was found to be the potent platelet inhibitor in healthy subjects while the citrus juices showed no effect [134]. Studies have shown that consumption of quercetin inhibits in vivo platelet activation, but the underlying mechanism is still unknown. Since collagen-induced platelet aggregation is associated with a burst of hydrogen peroxide, which in turn contributes to stimulating the phospholipase C pathway, one of the mechanisms could be the inhibition of collagen-induced hydrogen peroxide production, calcium mobilization, and 1, 3, 4-inositol triphosphate formation [135].
Quercetin interferes with many steps in eicosanoid metabolism. It inhibits phospholipase A2 activity [136] and blocks both the LOX137 and COX pathways of AA metabolism [138]. At micromolar concentrations (≤15-40 µM), quercetin inhibits COX-2 and LOX-5 activity [139]. Quercetin decreases platelet aggregation, increases platelet-derived NO release, and decreases superoxide production. In another study semi-synthesized quercetin derivatives--disodium Quercetin-7,4'-disulfate(DQD) inhibited pig platelet aggregation induced by thrombin. Its mechanism was attributed to its inhibitory effect on Ca2+ influx, intracellular Ca2+ mobilization, Ca2+/PL dependent PKC activity, and actin polymerization [140]. Previously, platelets were considered to be the main cell type involved in the pathogenesis of thrombosis, whereas only a minor role was attributed to leucocytes. However, many investigators recently realized that thrombosis involves multiple cells and the cell interactions further complicating this process [141] The interaction of platelet and leukocyte may be one of the key factors in the development of thrombotic diseases. Recently quercetin was reported to inhibit leukocyte aggregation and platelet-neutrophil adhesion with higher efficacy than aspirin. It is suggested that quercetin may be developed as an antithrombotic agent [142, 143]. However in a double-blind study [144], healthy men and women were assigned to either a quercetin-supplemented (1 g/d) group or a control group for 28 d. Plasma quercetin was 23-fold higher after quercetin supplementation but did not alter any of the thrombogenic risk factors. Similar observations were made in another randomized crossover study in which volunteers received 220 g onions/d (114 mg quercetin) as one of the treatments for 7 days [145]. In vitro, quercetin inhibits platelet aggregation [146] but the levels used ranged from 20 to 500 mmol/L of quercetin, which is several hundred times higher than the plasma levels reached in the human studies. Therefore, it is possible that the inhibitory effect of quercetin on platelet aggregation may require a certain minimum concentration in the plasma. It is also likely that quercetin elicits its cardioprotective effects via mechanisms other than thrombosis.
Inhibition of smooth muscle cell proliferation and migration
It is now apparent that ROS induce endothelial cell damage and VSMC growth which are responsible for cardiovascular remodeling, hypertension, atherosclerosis, heart failure, and restenosis [59, 147, 148].
Several lines of evidence indicate that ROS and mitogen-activated protein (MAP) kinases were involved in vascular remodeling under various pathological conditions. Recently, it was also reported that MAP kinases were sensitive to oxidative stress. MAP kinases play an important role in cell differentiation, growth, apoptosis, and the regulation of a variety of transcription factors and gene expressions. Ang II and endothelin-1 are the activators of MAP kinases in VSMC and are believed to cause vascular remodeling. Quercetin inhibited endothelin-1-induced proliferation of VSMC via the inhibition of JNK and p38, thereby suggesting the important role of ROS in MAP kinase activation and MAP kinase-mediated VSMC proliferation by endothelin70. During the formation of atherosclerotic lesions, the smooth muscle cells that line the coronary arteries multiply in number and begin to migrate into the interior of those vessels. When human aortic muscle cells are exposed to quercetin, this action is inhibited in a dose-dependent manner. Quercetin works by inhibiting the Mitogen-Activated Protein (MAP) kinase signaling pathway associated with smooth muscle cell migration. In another study, Japanese researchers found that quercetin inhibited the vascular smooth muscle cell hypertrophy seen during the development of coronary artery disease. They hypothesized that quercetin works by inhibiting the activation of a key MAP kinase called JNK(c-Jun N-terminal kinase ) by the angiotensin II enzyme. This in turn inhibits the protein synthesis necessary for proliferation and hypertrophy of smooth muscle cells [149, 150].
Quercetin is known to inhibit angiotensin II-induced hypertrophy and serum-induced smooth muscle cell proliferation. However, it is not known whether quercetin exerts similar cardioprotective effects in cells treated with TNF-alpha. Moon et al [151] investigated whether quercetin exerts the multiple suppressive effects on cytokine TNF-alpha-induced human aortic smooth muscle cells (HASMC) or not. Treatment of quercetin showed potent inhibitory effects on the DNA synthesis of cultured HASMC in the presence of TNF-alpha. These inhibitory effects were associated with reduced extracellular signal-regulated kinase (ERK)1/2 activity and G1 cell-cycle arrest. Treatment of quercetin, which induced a cell-cycle block in G1-phase, induced down-regulation of cyclins and CDKs and up-regulation of the CDK inhibitor p21 expression. Quercetin inhibited TNF-alpha-induced MMP-9 secretion on HASMC in a dose-dependent manner. This inhibition was characterized by down-regulation of MMP-9, which was transcriptionally regulated at NF-kappa B site and activation protein-1 (AP-1) site in the Matrix metallproteinkinase-9 (MMP-9 promoter). These findings indicate the efficacy of quercetin in inhibiting cell proliferation, G1- to S-phase cell-cycle progress, and MMP-9 expression through the transcription factors NF-kappaB and AP-1 on TNF-alpha-induced HASMC [151].
Anti-hypertensive action
Spanish researchers evaluated the antihypertensive effects of quercetin in an animal model of essential hypertension. Ten milligram per kilogram (10mg/kg) of quercetin given orally to spontaneously hypertensive rats for five weeks reduced systolic blood pressure by 18%, diastolic blood pressure by 23%, and mean arterial blood pressure by 21%. However quercetin had no effect on the normotensive rats. Quercetin was also found to decrease cardiac and renal hypertrophy, both of which follow hypertension and can lead to heart and kidney failure if left unchecked [152]. Quercetin shows vasodilator effects in isolated aortae stimulated with noradrenaline, KCl or phorbol esters and these effects are independent of the presence of endothelium Thus, this direct vasodilator effect might contribute to its antihypertensive effects [153, 154]. These effects were associated with a reduced oxidant status due to the antioxidant properties of the drug.
In another study quercetin (10 mg/kg) shows both antihypertensive and antioxidant properties in the deoxycorticosterone acetate-salt-hypertensive rats model, where verapamil (20 mg/kg/day) exhibits only antihypertensive effects. In this study, quercetin and verapamil treatments reduced systolic blood pressure of DOCA-salt rats in approximately 67.6 and 63.3% respectively. Both drugs reduced significantly hepatic and renal hypertrophy induced by DOCA-salt administration, while only quercetin prevented cardiac hypertrophy [155].
In another five weeks experimental study [156], quercetin treatment reduced systolic blood pressure of Goldblatt (GB) hypertensive rats. It also reduced cardiac hypertrophy and proteinuria developed in GB hypertensive rats. Normalisation of plasma nitrates plus nitrites (NOx) , thiobarbituric acid reactive substances (TBARS) concentrations and improvement of the antioxidant defences system in liver were also found to be accompanied with the antihypertensive effect of quercetin. Thus chronic oral treatment with quercetin shows both antihypertensive and antioxidant effects in this model of renovascular hypertension.
Support of mitochondrial function in cardiac cells
Inadequate blood supply to a region of the body for a certain period followed by the resumption of blood flow is termed Ischemia-reperfusion. Ischemia -reperfusion results in varying degrees of tissue damage depending on the duration and extent of the hypoperfusion. Myocardial damage induced by ischemia-reperfusion is due, at least in part, to the generation of ROS [157, 158]. There have been reports showing a close correlation between the production of ROS and simultaneous consumption of endogenous antioxidants [159, 160]. Indirect evidence consistent with this view is the cardioprotective effects of free radical scavengers and antioxidant supplements [161-163]. New research finds that rats given oral low dose of quercetin significantly protected against the injury that normally occurs during ischemia and reperfusion [164]. It has been established that mitochondria play a critical role in myocardial recovery from ischemia-reperfusion (I-R) damage, and in vitro experiments indicate that quercetin can exert a variety of direct effects on mitochondrial function. Quercetin treatment significantly decreased the impairment of cardiac function following I-R. This protective effect was associated with improved mitochondrial function after I-R. These results indicate that oral low dose quercetin is cardioprotective, possibly via a mechanism involving protection of mitochondrial function during I-R [165]. In another experimentally induced myocardial infarction in dogs by 60-minute occlusion of the coronary artery with the subsequent 24-hour reperfusion, the administration of quercetin solution (50 mg/kg) was found to improve the contractile function of the left ventricular myocardium, decrease the incidence of heart rate and conductivity disorders, limit the ischemic damage area, promote the preservation of the vessels' integrity, improve the coronary circulation and the prevent of intravascular thrombus formation [166]. Quercetin may also modulate ion channels, and possess structural similarities to several antiarrhythmic voltage-gated sodium channels (VGSC) inhibitors. By inhibiting cardiac voltage-gated sodium channels (VGSC), quercetin may contribute to cardioprotective and antiarrythmic effects [167].
Inhibition of NF-kappa B
The inflammatory mediator nuclear factor-kappa B (NF-κB), has gathered considerable attention in research circles because of its role in heart disease, kidney disease and other age-related degenerative disorders. One eye-opening study in mice compared the expression of NF-κB in regions of the coronary arteries that are more prone and less prone to the development of atherosclerotic plaques. They found that the NF-κB pathway is primed for activation in the atherosclerosis-prone arterial regions. A high-cholesterol diet activated NF-κB in precisely those regions, and increased expression of genes regulated by NF-κB which are involved in plaque formation and pathology [168]. Some previous data have indicated that quercetin has antiinflammatory property and a possible anti-inflammatory role of quercetin could be related to an interference with the NF-κB signaling pathway, which regulates the expression of various genes involved in inflammation. Recently Madhvan et al [169] demonstrated the inhibitory effects of quercetin on NF-κβ1 gene modulation. Their data suggested that the inhibitory effects of quercetin on TNF-α production may be mediated by down regulation of NF-κβ1 gene expression.
Reducing cardiovascular inflammation
Atherosclerosis is more and more being recognized as a chronic inflammatory process associated with enhanced serum levels of inflammation parameters, including in particular C-reactive protein. Cells found in early atherosclerotic lesions are typically inflammatory cells (monocytes/ macrophages and T-lymphocytes); and, there is convincing clinical and experimental evidence that formation of reactive oxygen species (ROS) is augmented during this chronic inflammatory process due to an imbalance between synthesis of ROS and neutralizing antioxidative defense mechanisms [170]. Thus the production of reactive oxygen species under pathological conditions may represent an important inflammatory trigger [171]. Inflammation not only promotes initiation and progression of atherosclerosis but also causes acute thrombotic complications of atherosclerosis. Novel therapeutic strategies specifically targeting inflammation thus bear great potential for the prevention and treatment of atherosclerotic vascular disease Secretions from mast cells play a central role in the development of inflammatory disorders, and recent research implicates them in cardiovascular inflammation, particularly following stress. Cardiovascular inflammation is now recognized as a key factor in coronary artery disease. Using a new and particularly sensitive blood test, known as high-sensitivity CRP, Ridker [172], has found that elevated levels of inflammation increase the risk of a heart attack by four and one-half times. That strong association makes CRP a far more accurate predictor of heart-attack risk than either cholesterol or homocysteine. Many studies have shown quercetin to inhibit mast cell secretion of inflammatory factors such as histamine, leukotrienes and prostaglandin D2 [173]. Several specific flavonoids may be helpful in reducing inflammation, specifically quercetin in a dose of 300-500 mg daily.
Quercetin obtained from numerous dietary sources like onions, applies, fruit juices, and tea is found to be a strong inhibitor of both COX-2 and 5-LOX enzymes involved in the production of the eicosanoids from arachidonic acid [138]. Thus the antioxidant property of Quercetin helps to neutralize free radicals, which promote inflammation.
CONCLUSION
Reactive oxygen species (ROS) contribute to the pathogenesis of cardiovascular diseases including hypertension, atherosclerosis, cardiac hypertrophy and heart failure. Oxidative stress results from excessive generation of ROS that outstrips the antioxidant system. By activating several signal transduction pathways, oxidative stress exerts noxious effects on cells of the vessel wall and leads to the initiation and progression of atherosclerosis culminating in acute events, such as acute coronary syndrome and stroke. Inhibition of ROS generation and function is a potential therapy to attenuate the extent of various cardiovascular diseases.
The hypothesis that dietary antioxidant quercetin lowers the risk of chronic disease has been developed from epidemiologic studies that consistently show that the consumption of whole foods, such as fruit and vegetables, is strongly associated with reduced risk of cardiovascular diseases. Therefore, it is reasonable for scientists to identify quercetin as a “magic bullet". Most of the cardioprotective actions of quercetin are thought to be due to its antioxidant properties such as metal chelators, free radical scavengers, and chain breaking antioxidants. Even though, one should recognize the complexity of interactions between pro-oxidants and antioxidants. A compound may act as an antioxidant in one oxidative challenge system and be neutral or even pro-oxidant in another, as seen in a study made by Myara et al [174], who demonstrated that quercetin can sequester and reduce the activity of oxidants inducing metals such as iron and copper. But with an anion exchange chromatographic method for testing potential inhibitors of low density lipoprotein oxidation, quercetin was found to have pro-oxidant activity. Altogether, the available evidence indicates that quercetin might be of therapeutic benefit in cardiovascular diseases even though prospective controlled clinical studies are still lacking.
REFERENCES
- World Health Organization. Preventing Chronic Disease a Vital Investment. Geneva: World Health Organization; 2005.
- Disease Control Priorities Project. Noncommunicable Diseases on the Rise in East, Central, and Southern Africa. retrieved from http://www.dcp2.org/file/68/DCPP_ECSA.pdf
- WHO Country Cooperation Strategy Republic of Mauritius 2004-2007 http://www.who.int/countryfocus/cooperation_strategy/ccs_mus_en.pdf
- Shuko NOJIRI, Hiroyuki DAIDA, Yutaka INABA. Antioxidants and Cardiovascular Disease: Still a Topic of Interest. Environ Health Prev. Med 2004;9:200-13.
- Noguchi N, Watanabe A, Shi H. Antioxidants in health and diseases: Antioxidant defense system. Free Rad Res. 2000;33:809-17.
- Hayes JD, McLellan LI. Glutathione and glutathione-dependent enzymes represent a co-ordinately regulated defence against oxidative stress. Free Radic Res. 1999;31:273-300.
- Rosenfeld ME. Inflammation, lipids, and free radicals: lessons learned from the atherogenic process. Semin Reprod Endocrinol. 1998;16:249-61.
- Hecht SS. Tobacco smoke carcinogens and lung cancer. J Natl Cancer Inst. 1999;91:1194-210.
- Ashok BT, Ali R. The aging paradox: free radical theory of aging. Exp Gerontol. 1999; 34:293-303.
- Stoclet JC, Chataigneau T, Ndiaye M, et al. Vascular protection by dietary polyphenols. Eur J Pharmacol. 2004 Oct 1;500(1-3):299-313.
- Manach C, Mazur A, Scalbert A. Polyphenols and prevention of cardiovascular diseases. Curr Opin Lipidol. 2005 Feb;16(1):77-84.
- Arts IC, Hollman PC. Polyphenols and disease risk in epidemiologic studies. Am J Clin Nutr. 2005 Jan;81(1 Suppl):317S-325S.
- Hubbard GP, Wolffram S, Lovegrove JA, et al. The role of polyphenolic compounds in the diet as inhibitors of platelet function. Proc Nutr Soc. 2003 May;62(2):469-78.
- Scalbert A, Manach C, Morand C, et al. Dietary polyphenols and the prevention of diseases. Crit Rev Food Sci Nutr. 2005;45(4):287-306.
- Chen J, Mehta JL. Role of Oxidative Stress in Coronary Heart Disease. Indian Heart J. 2004;56:163-73.
- Alexander RW. Theodore Cooper Memorial Lecture. Hypertension and the pathogenesis of atherosclerosis: oxidative stress and the mediation of arterial inflammatory response: a new perspective. Hypertension. 1995 Feb;25(2):155-61.
- Berliner JA, Heinecke JW. The role of oxidized lipoproteins in atherogenesis. Free Radic Biol Med. 1996;20:707-27.
- Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res. 2000;87:840-4.
- Suwaidi JA, Hamasaki S, Higano ST, et al. Long-term follow-up of patients with mild coronary artery disease and endothelial dysfunction. Circulation. 2000;101:948-54.
- Schachinger V, Britten MB, Zeiher AM. Prognostic impact of coronary vasodilator dysfunction on adverse long-term outcome of coronary heart disease. Circulation. 2000;101:1899-906.
- Halcox JP, Schenke WH, Zalos G, et al. Prognostic value of coronary vascular endothelial dysfunction. Circulation. 2002;106:653-8.
- Neunteufl T, Heher S, Katzenschlager R, et al. Late prognostic value of flow-mediated dilation in the brachial artery of patients with chest pain. Am J Cardiol. 2000;86:207-10.
- Perticone F, Ceravolo R, Pujia A, et al. Prognostic significance of endothelial dysfunction in hypertensive patients. Circulation. 2001;104:191-6.
- Heitzer T, Schlinzig T, Krohn K, et al. Endothelial dysfunction, oxidative stress, and risk of cardiovascular events in patients with coronary artery disease. Circulation. 2001;104:2673-8.
- Gokce N, Keaney JF Jr, Hunter LM, et al. Risk stratification for postoperative cardiovascular events via noninvasive assessment of endothelial function. Circulation. 2002 Apr 2;105(13):1567-72.
- Modena MG, Bonetti L, Coppi F, et al. Prognostic role of reversible endothelial dysfunction in hypertensive postmenopausal women. J Am Coll Cardiol. 2002;40:505-10.
- Lermon A, Burnett JC Jr. Intact and altered endothelium in regulation of vasomotion. Circulation. 1992;86(suppl III):III-12-II-19.
- Anderson TJ. Assessment and treatment of endothelial dysfunction in humans. J Am Coll Cardiol. 1999; 34:631-8.
- Herbert JM, Bono F, Savi P. The mitogenic effect of H2O2 for vascular smooth muscle cells is mediated by an increase of the affinity of basic fibroblast growth factor for its receptor. FEBS Lett. 1996;395:43-7.
- Ushio-Fukai M, Griendling KK, Becker PL, et al. Epidermal growth factor receptor transactivation by angiotensin II requires reactive oxygen species in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2001;21:489-95.
- Du J, Brink M, Peng T, et al. Thrombin regulates insulin-like growth factor-1 receptor transcription in vascular smooth muscle: characterization of the signaling pathway. Circ Res. 2001;88:1044-52.
- Delafontaine P, Ku L. Reactive oxygen species stimulate insulin-like growth factor I synthesis in vascular smooth muscle cells. Cardiovasc Res. 1997;33:216–22.
- Rocic P, Seshiah P, Griendling KK. Reactive oxygen species sensitivity of angiotensin II-dependent translation initiation in vascular smooth muscle cells. J Biol Chem. 2003;278: 36973-9.
- Peiro C, Lafuente N, Matesanz N, et al. High glucose induces cell death of cultured human aortic smooth muscle cells through the formation of hydrogen peroxide. Br J Pharmacol. 2001;133:967-74.
- Yorek MA, Dunlap JA. Effect of increased concentration of D-glucose or L-fucose on monocyte adhesion to endothelial cell monolayers and activation of nuclear factor-kappa B. Metabolism. 2002;51:225-34.
- Cheng JJ, Wung BS, Chao YJ, et al. Cyclic strain-induced reactive oxygen species involved in ICAM-1 gene induction in endothelial cells. Hypertension. 1998;31:125-30.
- Cominacini L, Garbin U, Pasini AF, et al. Antioxidants inhibit the expression of intercellular cell adhesion molecule-1 and vascular cell adhesion molecule-1 induced by oxidized LDL on human umbilical vein endothelial cells. Free Radic Biol Med. 1997;22:117-27.
- Zeng X, Dai J, Remick DG, et al. Homocysteine mediated expression and secretion of monocyte chemoattractant protein-1 and interleukin-8 in human monocytes. Cir Res. 2003;93:311-20.
- Burns AR, Bowden RA, Abe Y, et al. P-selectin mediates neutrophil adhesion to endothelial cell borders. J Leukoc Biol. 1999;65:299-306.
- Giannitsis E, Tettenborn I, Wiegand U, et al. Soluble L-selectin and neutrophil derived oxidative stress after pacing induced myocardial ischemia in chronic stable coronary artery disease. Pacing Clin Electrophysiol. 2000;23:309-14.
- Rattan V, Sultana C, Shen Y, et al. Oxidant stress-induced transendothelial migration of monocytes is linked to phosphorylation of PECAM-1. Am J Physiol. 1997;273:E453-E461.
- Li D, Chen H, Romeo F, et al. Statins modulate oxidized low-density lipoprotein-mediated adhesion molecule expression in human coronary artery endothelial cells: role of LOX-1. J Pharmacol Exp Ther. 2002;302:601-5.
- Rosenson RS, Brown AS. Statin use in acute coronary syndromes: cellular mechanisms and clinical evidence. Curr Opin Lipidol. 2002;13: 625-30.
- Ananyeva NM, Tjurmin AV, Berliner JA, et al. Oxidized LDL mediates the release of fibroblast growth factor-1. Arterioscler Thromb Vasc Biol. 1997;17: 445-53.
- Li D, Liu L, Chen H, et al. LOX-1 mediates oxidized low-density lipoprotein-induced expression of matrix metalloproteinases in human coronary artery endothelial cells. Circulation. 2003;107:612-7.
- Mehta JL, Hu B, Chen J, et al. Pioglitazone inhibits LOX-1 expression in human coronary artery endothelial cells by reducing intracellular superoxide radical generation. Arterioscler Thromb Vasc Biol. 2003;23:2203-8.
- Meilhac O, Zhou M, Santanam N, et al. Lipid peroxides induce expression of catalase in cultured vascular cells. J Lipid Res. 2000;41:1205-13.
- Rueckschloss U, Quinn MT, Holtz J, et al. Dose-dependent regulation of NAD(P)H oxidase expression by angiotensin II in human endothelial cells: protective effect of angiotensin II type 1 receptor blockade in patients with coronary artery disease. Arterioscler Thromb Vasc Biol. 2002; 22: 1845–51.
- Li DY, Zhang YC, Philips MI, et al. Upregulation of endothelial receptor for oxidized low-density lipoprotein (LOX-1) in cultured human coronary artery endothelial cells by angiotensin II type 1 receptor activation. Circ Res. 1999; 84:1043-9.
- Finkel T. Redox-dependent signal transduction. FEBS Lett. 2000;476:52–54.
- Patterson C, Madamanchi NR, Runge MS. The oxidative paradox: another piece in the puzzle. Circ Res. 2000;87:1074-6.
- Lo YY, Wong JM, Cruz TF. Reactive oxygen species mediate cytokine activation of c-Jun NH2-terminal kinases. J Biol Chem. 1996;271:15703-7.
- Watanabe Y, Suzuki O, Haruyama T, et al. Interferon-gamma induces reactive oxygen species and endoplasmic reticulum stress at the hepatic apoptosis. J Cell Biochem. 2003; 89: 244-53.
- Cheng TH, Cheng PY, Shih NL, et al. Involvement of reactive oxygen species in angiotensin II-induced endothelin-1 gene expression in rat cardiac fibroblasts. J Am Coll Cardiol. 2003; 42:1845-54.
- Cominacini L, Garbin U, Pasini AF, et al. Oxidized low-density lipoprotein increases the production of intracellular reactive oxygen species in endothelial cells: inhibitory effect of lacidipine. J Hypertens. 1998;16:1913-9.
- Colavitti R, Pani G, Bedogni B, et al. Reactive oxygen species as downstream mediators of angiogenic signaling by vascular endothelial growth factor receptor-2/KDR. J Biol Chem. 2002;277:3101-8.
- Sundaresan M, Yu ZX, Ferrans VJ, et al. Regulation of reactive-oxygen-species generation in fibroblasts by Rac1. Biochem J. 1996;318:379-82.
- Ishida M, Ishida T, Thomas SM, et al. Activation of extracellular signal-regulated kinases (ERK1/2) by angiotensin II is dependent on c-Src in vascular smooth muscle cells. Circ Res. 1998;82:7-12.
- Yoshizumi M, Kim S, Kagami S, et al. Effect of endothelin-1 on extracellular signal-regulated kinase and proliferation of human coronary artery smooth muscle cells. Br J Pharmacol. 1998;125:1019-27.
- Abe J, Kusuhara M, Ulevitch RJ, et al. Big mitogen-activated protein kinase 1 (BMK1) is a redox-sensitive kinase. J Biol Chem. 1996;271:16586-90.
- Ushio-Fukai M, Alexander RW, Akers M, et al. p38 Mitogen-activated protein kinase is a critical component of the redox-sensitive signaling pathways activated by angiotensin II. Role in vascular smooth muscle cell hypertrophy. J Biol Chem. 1998;273:15022-9.
- Chen J, Mehta JL, Haider N, et al. Role of caspases in Ox-LDL-induced apoptotic cascade in human coronary artery endothelial cells. Circ Res. 2004;94:370-6.
- Yoshizumi M, Tsuchiya K, Tamaki T. Signal transduction of reactive oxygen species and mitogen-activated protein kinases in cardiovascular disease. J Med Invest. 2001;48:11–24.
- Rodriguez-Puyol M, Griera-Merino M, Perez-Rivero G, et al. Angiotensin II induces a rapid and transient increase of reactive oxygen species. Antioxid Redox Signal. 2002;4:869–75.
- Gonzalez-Pacheco FR, Caramelo C, Castilla MA, et al. Mechanism of vascular smooth muscle cells activation by hydrogen peroxide: role of phospholipase C gamma. Nephrol Dial Transpantl. 2002;17:392-8.
- Roy S, Parinandi N, Zeigelstein R, et al. Hyperoxia alters phorbol ester-induced phospholipase D activation in bovine lung microvascular endothelial cells. Antioxid Redox Signal. 2003;5:217–28.
- Tarpey MM, Fridovich I. Methods of detection of vascular reactive species: nitric oxide, superoxide, hydrogen peroxide, and peroxynitrite. Cir Res. 2001;89:224-36.
- Brennan LA, Black SM. Nitric oxide activates p21ras and leads to the inhibition of endothelial NO synthase by protein nitration. DNA Cell Biol. 2003;22:317-28.
- Spiecker M, Darius H, Liao JK. A functional role of I kappa B-epsilon in endothelial cell activation. J Immunol. 2000;164: 3316-22.
- Kyaw M, Yoshizumi M, Tsuchiya K, et al. Antioxidants inhibit JNK and p38 MAPK activation but not ERK 1/2 activatio by angiotensin II in rat aortic smooth muscle cells. Hypertens Res. 2001;24:251-61.
- Richard DE, Berra E, Pouyssegur J. Nonhypoxic pathway mediates the induction of hypoxia-inducible factor 1 alpha in vascular smooth muscle cells. J Biol Chem. 2000;275:26765-71.
- Wung BS, Cheng JJ, Chao YJ, et al. Modulation of Ras/ Raf/extracellular signal-regulated kinase pathway by reactive oxygen species is involved in cyclic strain-induced early growth response-1 gene expression in endothelial cells. Cir Res. 1999;84:804-12.
- Matkovics A. Antioxidants and vascular diseases. Orv Hetil. 2003;144:475-81.
- Meng CQ, Somers PK, Rachita CL, et al. Novel phenolic antioxidants as multifunctional inhibitors of inducible VCAM-1 expression for use in atherosclerosis. Bioorg Med Chem Lett. 2002;12:2545-8.
- Semb AG, van Wissen S, Ueland T, et al. Raised serum levels of soluble CD40 ligand in patients with familial hypercholesterolemia: down regulatory effect of statin therapy. J Am Coll Cardiol. 2003;41:275-9.
- Tsiara S, Elisaf M, Mikhailidis DP. Early vascular benefits of statin therapy. Curr Med Res Opin. 2003;19:540-56.
- Bonetti PO, Lerman LO, Napoli C, et al. Statin effects beyond lipid lowering-Are they clinically relevant? Eur Heart J. 2003;24:225-48.
- Mehta JL, Li DY, Yang H, et al. Angiotensin II and IV stimulate expression and release of plasminogen activator inhibitor-1 i cultured human coronary artery endothelial cells. J Cardiovasc Pharmacol. 2002;39:789-94.
- Li D, Chen H, Mehta JL. Angiotensin II via activation of type 1 receptor upregulates expression of endoglin in human coronary artery endothelial cells. Hypertension. 2001;38:1062-7.
- Inoue I, Goto S, Matsunaga T, et al. The ligands/activators for peroxisome proliferator-activated receptoralpha (PPAR alpha) and PPAR gamma increase Cu2+, Zn2+- superoxide dismutase and decrease p22phox message expressions in primary endothelial cells. Metabolism. 2001;50:3-11.
- Braunstein S. Cardiovascular disease and benefits of thiazolidinediones. Postgrad Med. 2003;99:45-52.
- Varo N, Vicent D, Libby P, Nuzzo R, et al. Elevated plasma levels of the atherogenic mediator soluble CD40 ligand in diabetic patients: a novel target of thiazolidinediones. Circulation. 2003;107:2664-9.
- Wang M, Tafuri S. Modulation of PPAR gamma activity with pharmaceutical agents: treatment of insulin resistance and atherosclerosis. J Cell Biochem. 2003;89:38–47.
- Tao L, Liu HR, Gao E, et al. Antioxidative, antinitrative, and vasculoprotective effects of a peroxisome proliferator-activated receptor-gamma agonist in hypercholesterolemia. Circulation. 2003;108:2805-11.
- Chen J, Li D, Mehta JL. Tumor necrosis factor-α-induced apoptosis of human coronary artery endothelial cells: modulation by the peroxisome proliferator-activated receptor-gamma ligand pioglitazone. J Cardiovasc Pharmacol Ther. 2004;9:35-41.
- Viberti GC. Rosiglitazone: potential beneficial impact on cardiovascular disease. Int J Clin Pract. 2003;57:128-34.
- Meagher E, Rader DJ. Antioxidant therapy and atherosclerosis: animal and human studies. Trends Cardiovasc Med. 2001;11:162-5.
- Mehta JL, Mehta J. Antioxidants and vitamins in your cardiac patient: are they helpful? Cardiol Rev. 1999;7:56-61.
- Engler MM, Engler MB, Malloy MJ, et al. Antioxidant vitamins C and E improve endothelial function in children with hyperlipidemia: Endothelial Assessment of Risk from Lipids in Youth (EARLY) trial. Circulation. 2003;108:1059-63.
- Renaud S, de Lorgeril M. Wine, alcohol, platelets, and the French paradox for coronary heart disease. Lancet. 1992;339:1523-6.
- Covas MI, Fito M, Marrugat J, et al. Coronary disease protective factors: antioxidant effect of olive oil. Therapie. 2001 Sep-Oct;56(5):607-11.
- Serra-Majem L, Ribas L, Treserras R et al. How could changes in diet explain changes in coronary heart disease mortality in Spain? The Spanish paradox. Am J Clin Nutr. 1995;61(s):1351-9.
- Mata P, Alonso R, Lopez-Farre A et al. Effect of dietary fat saturation on LDL oxidation and monocyte adhesion to human endothelial cells in vitro. Arteriosc Thromb Vasc Biol. 1996;16:1347-55.
- Nicolaiew N, Lemort N, Adorni L. et al. Comparison between extra-virgin olive oil and oleic acid rich sunflower oil: Effects on postprandial lipemia and LDL susceptibility to oxidation. Ann Nutr Metab. 1998;42:251-60.
- Ramirez-Tortosa MC,Urbano G, Lopez-Jurado M et al. Extra-virgin olive oil increases the resistance of LDL to oxidation more than refined olive oil in free living men with peripheral vascular disease. J Nutr. 1999;129:2177-83.
- Liu S, Manson JE, Lee IM, et al. Fruit and vegetable intake and risk of cardiovascular disease: the Women’s Health Study. Am J Clin Nutr. 2000;72:922-8.
- Joshipura KJ, Hu FB, Manson JE, et al. The effect of fruit and vegetable intake on risk of coronary heart disease. Ann Int Med. 2001;134:1106-14.
- Liu S, Lee I M, Ajani U, Cole, et al. Intake of vegetables rich in carotenoids and risk of coronary heart disease in men:The Physicians’ Health Study. Intl J Epidermiology. 2001; 30:130-5.
- van’t Veer P, Jansen MC, Klerk M,et al. Fruits and vegetables in the prevention of cancer and cardiovascular disease. Public Health Nutr. 2000;3:103-7.
- Hertog MGL, Feskens EJM, Hollman PCH, et al. Dietary antioxidant flavonoids and risk of coronary heart disease: the Zutphen Elderly study. Lancet. 1993;342:1007-11.
- Huxley RR, Neil HAW. The relation between dietary flavonol intake and coronary heart disease mortality: a meta-analysis of prospective cohort studies. Europ J Clin Nutr. 2003;57:904-8.
- Beecher GR. Overview of dietary flavonoids: nomenclature, occurrence and intake. J Nutr. 2003;133:3248S–3254S.
- Chopra M, Fitzsimons PEE, Strain JJT, et al. Nonalcoholic red wine extract and quercetin inhibit LDL oxidation without affecting plasma antioxidant vitamin and carotenoid concentrations. Clin Chem. 2000;46:1162-70.
- Fuhrman B, Aviram M. Flavonoids protect LDL from oxidation and attenuate atherosclerosis. Curr Opin Lipidol. 2001;12:41-8.
- Caltagirone S, Ranelletti FO, Rinelli A, et al. Interaction with type II estrogen binding sites and antiproliferative activity of tamoxifen and quercetin in human non-small-cell lung cancer. Am J Respir Cell Mol Biol. 1997;17:51-9.
- Pereira MA, Grubbs CJ, Barnes LH, et al. Effects of the phytochemicals, curcumin and quercetin, upon azoxymethane-induced colon cancer and 7, 12-dimethylbenz[a]anthracene-induced mammary cancer in rats. Carcinogenesis. 1996;17:1305-11.
- Siess MH, Leclerc J, Canivenc-Lavier MC, et al. Heterogenous effects of natural flavonoids on monooxygenase activities in human and rat liver microsomes. Toxicol Appl Pharmac. 1995;130:73-8.
- Conseil G, Baubichon-Cortay H, Dayan G, et al. Flavonoids: a class of modulators with bifunctional interactions at vicinal ATP- and steroid-binding sites on mouse P-glycoprotein. Proc Natl Acad Sci. 1998;95:9831-6.
- Agullo G, Gamet-Payrastre L, Manenti S, et al. Relationship between flavonoid structure and inhibition of phosphatidylinositol 3-kinase: a comparison with tyrosine kinase and protein kinase C inhibition. Biochem Pharmac. 1997;53:1649-57.
- Formica JV, Regelson W. Review of the biology of quercetin and related bioflavonoids. Food Chem Toxicol. 1995;33:1061-80.
- Engelhardt U, Finger A, Herzig B, et al. Determination of flavonol glycosides in black tea. Deutsche Lebensmittel-Rundschau. 1992;88:69-73.
- Kiviranta J, Huovinen K, Hiltunen R. Variation in phenolic substances in onion. Acta Pharmaceutica Fennica. 1988;97:67-72.
- Hollman P. Buysman MP, van Gameren Y, et al. The sugar moiety is a major determinant of the absorption of dietary flavonoid glycosides in man. Free Rad Res. 1999;31:569-73.
- Hollman PC, van Trijp J, Buysman MN, et al. Relative bioavailability of the antioxidant flavonoid quercetin from various foods in man. FEBS Lett. 1997;418:152-6.
- Hollman PC, de Vries JH, van Leeuwen SD, et al. Absorption of dietary quercetin glycosides and quercetin in healthy ileostomy volunteers. Am J Clin Nutr. 1995;62:1276-82.
- Manach C, Morand C, Crespy V, et al. Quercetin is recovered in human plasma as conjugated derivatives which retain antioxidant properties. FEBS Lett. 1998;426:331-6.
- Hollman PCH, Katan M B. Absorption, metabolism and bioavailability of flavonoids. Flavonoids in Health and Disease. Rice Evans C. Packer L. eds. Marcel Dekker Inc New York; 1998:483-522.
- Parthasarathy S, Steinberg D, Witztum JL. The role of oxidized low-density lipoproteins in the pathogenesis of atherosclerosis. Ann Rev Med. 1992;43:219-25.
- Navab M, Imes S, Hama S, et al. Monocyte transmigration induced by modifications of LDL in cocultures of human aortic wall cells is due to induction of monocyte chemotactic protein I synthesis and is abolished by HDL. J Clin Invest. 1991;88:2039-46.
- Kugiyama K, Kerns SA, Morrisett JD, et al. Impairment of endothelial-dependent arterial relaxation by lysolecithin in modified low-density lipoproteins. Nature. 1990;344:160-2.
- Kita T, Kume N, Ishii K, et al. Oxidized LDL and expression of monocyte adhesion molecules. Diabetes Res Clin Pract. 1999;45:123-6.
- Huang YH, Schafer-Elinder L, Wu R, et al. Lysophosphatidylcholine (LPC) induces proinflammatory cytokines by a platelet-activating factor (PAF) receptor-dependent mechanism. Clin Exp Immunol. 1999;116:326-31.
- Carolina Aliaga, Eduardo A. Lissi. Comparison of the free radical scavenger activities of quercetin and rutin — An experimental and theoretical study. Canadian Journal of Chemistry. 2004;82(12):1668-73.
- Sim GS, Lee BC, Cho HS, et al. Structure activity relationship of antioxidative property of flavonoids and inhibitory effect on matrix metalloproteinase activity in UVA-irradiated human dermal fibroblast. Arch Pharm Res. 2007 Mar;30(3):290-8.
- da Silva EL, Abdalla DS, Terao J. Inhibitory effect of flavonoids on low-density lipoprotein peroxidation catalyzed by mammalian 15-lipoxygenase. IUBMB life. 2000 Apr;49(4):289-95.
- Chaillou LL, Nazareno MA. New method to determine antioxidant activity of polyphenols. J Agric Food Chem. 2006 Nov;54(22):8397-402.
- Terao J, Piskula MK. Flavonoids and membrane lipid peroxidation inhibition. Nutrition. 1999;15:790-1.
- Robak J, Gryglewski RJ. Flavonoids are scavengers of superoxide anion. Biochem Pharmacol. 1988;37:83-8.
- Husain SR, Cillard J, Cillard P. Hydroxy radical scavenging activity of flavonoids. Phytochemistry. 1987;26:2489-92.
- Sorata Y, Takahama U, Kimura M. Protective effect of quercetin and rutin on photosensitized lysis of human erythrocytes in the presence of hematoporphyrin. Biochem Biophys Acta. 1982;799:313-17.
- Afanas'ev IB, Dorozhko AI, Brodskii AV, et al. Chelating and free radical scavenging mechanisms of inhibitory action of rutin and quercetin in lipid peroxidation. Biochem Pharmacol. 1989 Jun;38(11):1763-9.
- Gryglewski RJ, Korbut R, Robak J, et al. Mechanism of antithrombotic action of flavonoids. Biochem Pharmacol. 1987;36:317-22.
- Osman HE, Maalej N, Shanmuganayagam D, et al. Grape juice but not orange or grapefruit juice inhibits platelet activity in dogs and monkeys. J Nutr. 1998;128:2307-12.
- Keevil J, Osman H, Maalej N, et al. Grape juice, but not orange juice or grapefruit juice, inhibits human platelet aggregation. J Nutr. 2000 Jan;130(1):53-6.
- Pignatelli P, Pulcinelli FM, Celestini A, et al. The flavonoids quercetin and catechin synergistically inhibit platelet function by antagonizing the intracellular production of hydrogen peroxide. Am J Clin Nutr. 2000 Nov; 72(5):1150-5.
- Havsteen B. Flavonoids, a class of natural products of high pharmacological potency. Biochem Pharmacol. 1983;32:1141-8.
- Baumann J, Bruchhausen F, Wurm G. Flavonoids and related compounds as inhibition of arachidonic acid peroxidation. Prostaglandins. 1980;20:627-39.
- Welton AF, Hurley J, Will P. Flavonoids and arachidonic acid metabolism. Prog Clin Biol Res. 1988;280:301-12.
- Welton AF, Tobias LD, Fiedler-Nagy C, et al. Effect of flavonoids on arachidonic acid metabolism. Prog Clin Biol Res. 1986;213:231-42.
- Freedman JE, Parker C, Li L, et al. Select flavonoids and whole juice from purple grapes inhibit platelet function and enhance nitric oxide release. Circulation. 2001 Jun 12;103(23):2792-8.
- Marcus AJ, Safier LB. Thromboregulation: multicellular modulation of platelet reactive discourse in hemostasis and thrombosis. FASEB J. 1993;7(6):516-22.
- Li FG, Wang ZY, Ruan CG, et al. The effects of quercetin on HLA f unction and metabolism. Chin Pharm Bull. 1991;7(6):466-9.
- Liu W, Liang NC. Inhibitory effect of disodium quercetin-7,4'-disulfate on aggregation of pig platelets induced by thrombin and its mechanism. Acta Pharmacol Sin. 2000 Aug;21(8):737-41.
- Conquer JA, Maini G, Azzini E, et al. Supplementation with quercetin markedly increases plasma quercetin concentration without effect on selected risk factors for heart disease in healthy subjects. J Nutr. 1998;128:593–7.
- Janssen K, Mensink RP, Cox FJJ, et al. Effects of the flavonoids quercetin and apigenin on hemostasis in healthy volunteers: results from an in vitro and a dietary supplement study. Am J Clin Nutr. 1998;67:255–62.
- Tzeng SH, Ko WC, Ko FN, et al. Inhibition of platelets by some flavonoids. Thromb Res. 1991;64:91–100.
- Abe J, Berk BC. Reactive oxygen species as mediators of signal transduction in cardiovascular disease. Trends Cardiovasc Med. 1998;8:59-64.
- Griendling KK, Sorescu D, Ushio-Fukai M. NAD(P)H oxidase: role in cardiovascular biology and disease. Circ Res. 2000;86:494-501.
- Alcocer F, Whitley D, Salazar Gonzales JF, et al. Quercetin inhibits human vascular smooth muscle cell proliferation and migration. Surgery. 2002;131:198-204.
- Yoshizumi M, Tsuchiya K, Kirima K, et al. Quercetin inhibits Shc- and phosphatidylinositol 3-kinase-mediated c-jun N-terminal kinase activation by angiotensin II in cultured rat aortic smooth muscle cells. Mol Pharmacol. 2001;60:656-65.
- Moon SK, Cho GO, Jung SY, et al. Quercetin exerts multiple inhibitory effects on vascular smooth muscle cells: role of ERK1/2, cell-cycle regulation, and matrix metalloproteinase-9. Biochem Biophys Res Commun. 2003 Feb 21;301(4):1069-78.
- Duarte J, Pérez-Palencia R, Vargas F, et al. Antihypertensive effects of the flavonoid quercetin in spontaneously hypertensive rats. Br J Pharmacol. 2001;133:117-24.
- Duarte J, Perez-Vizcaino F, Utrilla P, et al. Vasodilatory effects of flavonoids in rat aortic smooth muscle. Structure-activity relationships. Gen Pharmacol. 1993;24:857-64.
- Duarte J, Pérez-Vizcaíno F, Zarzuelo A, et al. Vasodilator effects of quercetin in isolated rat vascular smooth muscle. Eu. J Pharmacol. 1993;239:1-7.
- Galisteo M, García-Saura MF, Jiménez R, et al. Effects of chronic quercetin treatment on antioxidant defence system and oxidative status of deoxycorticosterone acetate-salt-hypertensive rats. Molecular and cellular biochemistry. 2004;(259):91-9.
- Garcia-Saura MF, Galisteo M, Villar IC, et al. Effects of chronic quercetin treatment in experimental renovascular hypertension. Mol Cell Biochem. 2005 Feb;270(1-2):147-55.
- Kloner RA, Przyklenk K, Whittaker P. Deleterious effects of oxygen radicals in Ischemia –reperfusion. Resolved and unresolved issues. Circulation. 1989;80(5):1115-27.
- Kilgore KS, Lucchesi BR. Reperfusion injury after myocardial infarction: the role of free radicals and the inflammatory response. Clin Biochem. 1993;26(5):359-70.
- Ko KM, Garnett ME, Godin DV. Altered antioxidant status in ischemic/reperfused rabbit myocardium: reperfusion time-course study. Can J Cardiol. 1990;6(7):299-304.
- Leichtweis S, Ji LL. Glutathione deficiency intensifies ischaemia-reperfusion induced cardiac dysfunction and oxidative stress. Acta Physiol Scand. 2001;172(1):1-10.
- Steare SE, Yellon DM. The potential for endogenous myocardial antioxidants to protect the myocardium against ischaemia-reperfusion injury: refreshing the parts exogenous antioxidants cannot reach? J Mol Cell Cardiol. 1995;27(1):65-74.
- Alberola A, Such L, Gil F, et al. Protective effect of N- acetylcysteine on ischaemia-induced myocardial damage in canine heart. Naunyn Schmiedebergs Arch Pharmacol. 1991;343(5):505-10.
- Nishinaka Y, Sugiyama S, Yokota M, et al. The effects of a high dose of ascorbate on ischemia-reperfusion-induced mitochondrial dysfunction in canine hearts. Heart Vessels. 1992;7(1):18-23.
- Cui J, Cordis GA, Tosaki A, et al. Reduction of Myocardial Ischemia Reperfusion Injury with Regular Consumption of Grapes. Annals of the New York Academy of Sciences. 2002;957:302-7.
- Brookes PS, Digerness SB, Parks DA, et al. Mitochondrial function in response to cardiac ischemia-reperfusion after oral treatment with quercetin. Free Rad Biol Med. 2002 jun;32(11):1220-8.
- Kolchin IuN, Maksiutina NP, Balanda PP, et al. The cardioprotective action of quercetin in experimental occlusion and reperfusion of the coronary artery in dogs. Farmakol Toksikol. 1991 Nov-Dec;54(6):20-3.
- Wallace CH, Baczkó I, Jones L, et al. Inhibition of cardiac voltage-gated sodium channels by grape polyphenols. Br J Pharmacol. 2006 Nov;149(6):657-65.
- Hajra L, Evans AI, Chen M, et al. The NF-kappa B signal transduction pathway in aortic endothelial cells is primed for activation in regions predisposed to atherosclerotic lesion formation. Proc Natl Acad Sci U S A. 2000 Aug;97(16):9052-7.
- Nair MP, Mahajan S, Reynolds JL, et al. The Flavonoid Quercetin Inhibits Proinflammatory Cytokine (Tumor Necrosis Factor Alpha) Gene Expression in Normal Peripheral Blood Mononuclear Cells via Modulation of the NF-κβ System. Clin Vaccine Immunol. 2006 March;13(3):319-28.
- Galle J. Atherosclerosis and arteriitis: implications for therapy of cardiovascular disease. Herz. 2004 Feb;29(1):4-11.
- Sudano I, Spieker LE, Hermann F, et al. Protection of endothelial function: targets for nutritional and pharmacological interventions. J Cardiovasc Pharmacol. 2006;47 (Suppl 2):S136-50; discussion S172-6.
- Ridker PM, Hennekens CH, Buring JE, et al. C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N Engl J Med. 2000 Mar 23;342(12):836-43.
- Kimata M, Shichijo M, Miura T, et al. Effects of luteolin, quercetin and baicalein on immunoglobulin E-mediated mediator release from human cultured mast cells. Clin Exp Allergy. 2000 Apr;30(4):501-8.
- Myara I, Pico I, Vedie B, et al. A method to screen for the antioxidant effect of compounds on low-density lipoprotein (LDL): illustration with flavonoids. J Pharmacol Toxicol Methods. 1993 Oct;30(2):69-73.